Autores
- Jesús, Florencia (1); Pequeño, Fiamma (2); Gérez-García, Natalia (3), Niell, Silvina (1); Cesio, María Verónica (1, 2, 3); Heinzen, Horacio (2, 3)
- PDU "ABORDAJE HOLÍSTICO AL IMPACTO DE AGROQUÍMICOS SOBRE ALIMENTOS Y AMBIENTE", GACT, CENUR Litoral Norte, Universidad de la República, Paysandú, Uruguay.
- Departamento de Química del Litoral, GACT, CENUR Litoral Norte, Universidad de la República, Paysandú, Uruguay.
- Cátedra de Farmacognosia y Productos Naturales, Departamento de Química Orgánica, GACT, Universidad de la República, Montevideo, Uruguay.
El uso de la miel como alimento natural establece altas exigencias en cuanto a su calidad. Dado que la producción de miel se realiza de la mano con las actividades agrícolas, se han detectado residuos de pesticidas en mieles en varios países, y Uruguay es uno de ellos, a veces excediendo los límites máximos de residuos (LMR). Este trabajo presenta el ajuste para la determinación de glifosato en miel. Se empleó una metodología moderna de extracción con metanol, denominada QuPPe, desarrollada específicamente para el análisis de pesticidas polares empleando cromatografía líquida de intercambio iónico acoplada a un detector de masas en tándem (LC–MS/MS). Por las particularidades de la molécula de glifosato, no puede ser incluido en un método multirresiduo. El método evaluado y validado fue desarrollado por el Laboratorio de Referencia Europeo para el análisis monorresiduo de pesticidas (EURL–SRM). Para su validación, se siguieron las directrices del documento SANTE y se obtuvieron resultados aceptables incluso al LMR establecido por la Unión Europea (0,050 mg kg-1). Se analizaron muestras reales uruguayas y en algunos casos se encontraron residuos de glifosato. La metodología es útil para el control de calidad e inocuidad de la miel en lo que a residuos de glifosato respecta.
Palabras clave: Pesticidas polares, extracción con metanol, cromatografía líquida de intercambio iónico; espectrometría de masas en tándem.
Introducción
La miel es el producto de la colmena más consumido por el ser humano, por lo que debe tener asegurada su inocuidad alimentaria. Los pesticidas, posibles contaminantes de la miel, pueden ingresar a la colmena a través de varias rutas; por ejemplo: el mal uso de los agroquímicos empleados para la sanitización de la colmena, la contaminación ambiental, o el no cumplimiento de las buenas prácticas agrícolas (BPA). El uso de la miel como alimento natural establece altas exigencias en cuanto a su calidad, pero la producción de miel se realiza de la mano con las actividades agrícolas. La contaminación de las abejas y de los productos de la colmena (miel, cera y polen) tiene implicancias tanto económicas como sanitarias. Dado que el sistema nervioso de la abeja es afectado por dosis subletales de agroquímicos, alterando los parámetros biológicos de funcionamiento general de la colmena se ve disminuida su productividad y amenazada su sobrevivencia (Decourtye y Pham-Delègue, 2002; Decourtye, et al., 2005; Decourtye, et al., 2011; El Hassani, et al., 2008; Smodiš Škerl, et al., 2010). Para poder asegurar que la miel es un alimento inocuo, es importante el análisis de la miel con el objetivo de control de los residuos de pesticidas en este alimento, tanto para consumo interno como para exportación. En Uruguay, la producción nacional se comercializa en un 82% en el exterior y el 18% restante se destina al mercado interno y cubre casi la totalidad de la demanda (DIGEGRA, 2018).
Recientemente, han existido problemas de comercialización de mieles uruguayas en países de la Unión Europea, Alemania por ejemplo, a raíz de la presencia de glifosato en concentraciones superiores a las permitidas, lo que provocó el rechazo de partidas de exportación y afectó negativamente a la apicultura uruguaya. El límite máximo de residuos (LMR) de glifosato en miel está definido en la Unión Europea como 0,050 mg kg-1 (Unión Europea, 2013), aunque el valor puede variar de acuerdo a reglamentaciones internas de cada país dentro de Europa, que deben tenerse en cuenta en caso de exportación.
El glifosato (N-fosfonometilglicina) es uno de los herbicidas más ampliamente utilizados en todo el mundo para el control de malezas tanto en agricultura como en áreas urbanas. Es un herbicida de amplio espectro, no selectivo, sistémico y de post-emergencia. Su mecanismo de acción consiste en interferir en la biosíntesis de los aminoácidos aromáticos fenilalanina, tirosina y triptófano, mediante la inhibición de la enzima 5-enolpiruvilshikimato-3-fosfato sintasa (EPSPS). Los aminoácidos aromáticos son necesarios para el metabolismo primario y secundario de las plantas, y una deficiencia en ellos impide su crecimiento y les provoca la muerte. La enzima EPSPS no está presente en mamíferos, quienes obtienen sus aminoácidos esenciales a partir de la dieta.
La molécula de glifosato es relativamente simple: consiste en la unión del aminoácido glicina con un grupo fosfonometil (Figura 1). Es una molécula pequeña, muy polar, que no posee grupos cromóforos; es poco volátil, con gran solubilidad en agua y baja en solventes orgánicos.
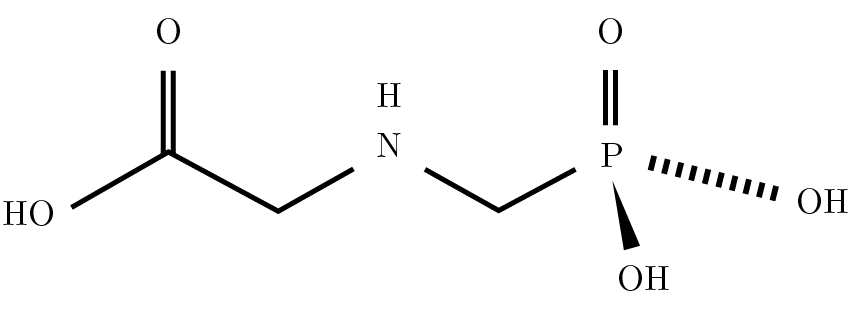
Figura 1. Estructura de la molécula de glifosato (PM = 169).
Debido a las particularidades de este compuesto, no es posible analizarlo mediante una metodología multirresiduo, es decir, simultáneamente con otros pesticidas. Las metodologías más utilizadas para su análisis consisten en la derivatización de la molécula de glifosato, por ejemplo, con el reactivo 9–fluorenilmetilcloroformato (FMOC–Cl), con el fin de aumentar su masa y hacerla más apolar para su posterior determinación por cromatografía gaseosa o líquida utilizando columnas cromatográficas y/o detectores convencionales (Gill, et al., 2017). Sin embargo, el uso de reacciones de derivatización en la preparación de la muestra implica mayores tiempos de preparación de muestra y de análisis. Además, en el caso del FMOC–Cl pueden producirse subproductos que potencialmente dañen el instrumental. Por lo tanto, la posibilidad de contar con un método de preparación de muestra más sencillo cobra importancia cuando se desea implementar el análisis rutinario de este tipo de compuestos, por ejemplo. Ha sido desarrollada por los laboratorios de referencia de la Unión Europea (EURL–SRM) una metodología para el análisis de varios pesticidas muy polares (método QuPPe, del inglés Quick method for the analysis of numerous highly Polar Pesticides) en alimentos de origen vegetal mediante cromatografía líquida acoplada a espectrometría de masas en tándem (LC–MS/MS) (Anastassiades, et al., 2017). Este método utiliza una preparación de muestra sencilla con equipamientos específicos y columnas especiales de intercambio iónico. Las desventajas que presenta son el tiempo empleado para estabilizar las condiciones de trabajo de la columna previo a su uso y la necesidad de aumentar la frecuencia de limpieza y mantenimiento del espectrómetro de masas debido al tipo de fase móvil utilizado.
En este trabajo se muestra el ajuste y posterior validación del método QuPPe bajo las condiciones del laboratorio del Grupo de Análisis de Compuestos Traza (GACT) de la Universidad de la República, para poder dar respuesta a los pedidos de los apicultores de una metodología aplicable de rutina.
Materiales y métodos
Reactivos y estándares
Se utilizaron metanol y acetonitrilo de calidad HPLC de Pharmco – Apper (Brookfield, CT, USA) y de Merck (Darmstadt, Germany), respectivamente. El agua (H2O) fue desionizada en el laboratorio con un purificador de agua Thermo Scientific (Marietta, OH, USA) EASYpure RoDi. El ácido fórmico p.a. (98 – 100%) fue obtenido de Merck y una solución de ácido fórmico al 1% (v/v) fue preparada en metanol. El ácido cítrico monohidrato p.a. (pureza 99,7%) y la dimetilamina (DMA) anhidro (> 99%) utilizados fueron de BDH Chemicals Ltd. (Poole, England) y de Sigma-Aldrich (St. Louis, MO, USA), respectivamente. El estándar analítico de glifosato, sólido cristalino de pureza 98%, fue suministrado por Dr. Ehrenstorfer (Augsburg, Germany). La solución stock del estándar se preparó en matraz aforado a una concentración de 0,2 mg mL-1 utilizando como disolvente una mezcla 1:1 de H2O y ácido fórmico 1% (v/v) en metanol, considerando la pureza del estándar sólido, el cual se pesó en balanza analítica. Las soluciones de trabajo se prepararon diluyendo apropiadamente la solución stock con acetonitrilo 10% (v/v) en H2O. Todas las soluciones se conservaron en la oscuridad a menos de 4 °C.
Instrumentos
Se trabajó con pipetas automáticas adecuadas para medir volúmenes de 10–100 µL, 20–200 μL, 100–1000 μL y 1–10 mL de Socorex (Lausanne, Switzerland), balanzas analíticas con capacidad de medir hasta 0,01 mg o 10 mg de Shimadzu (Kyoto, Japan). Los tubos con tapa a rosca de polipropileno descartables para centrífuga de 50 mL (30 × 115 mm) fueron adquiridos de Kartell (Milan, Italy). El vortex utilizado para la homogenización de las muestras de miel fue un VM–10 de Daihan Scientific Co. (Seúl, Corea). La centrífuga, capaz de alcanzar al menos 3000 × g, fue una SL16 de Thermo Electron (Langenselbold, Germany). El pH–metro utilizado para ajustar el pH de la fase móvil B fue un pH Tutor Oakton® fabricado por Eutch Instruments (Singapore). Los viales de autosampler de 1,5 mL fueron suministrados por Thermo Scientific (Rockwood, TN, USA). Se utilizaron insertos de polipropileno de 0,25 mL de Waters Corporation (Milford, MA, USA). Filtros para jeringa de PVDF (13 mm, 0,45 µm) fueron adquiridos de Lubitech Technologies (Songjiang, Shanghai, China).
Condiciones experimentales de LC–MS/MS
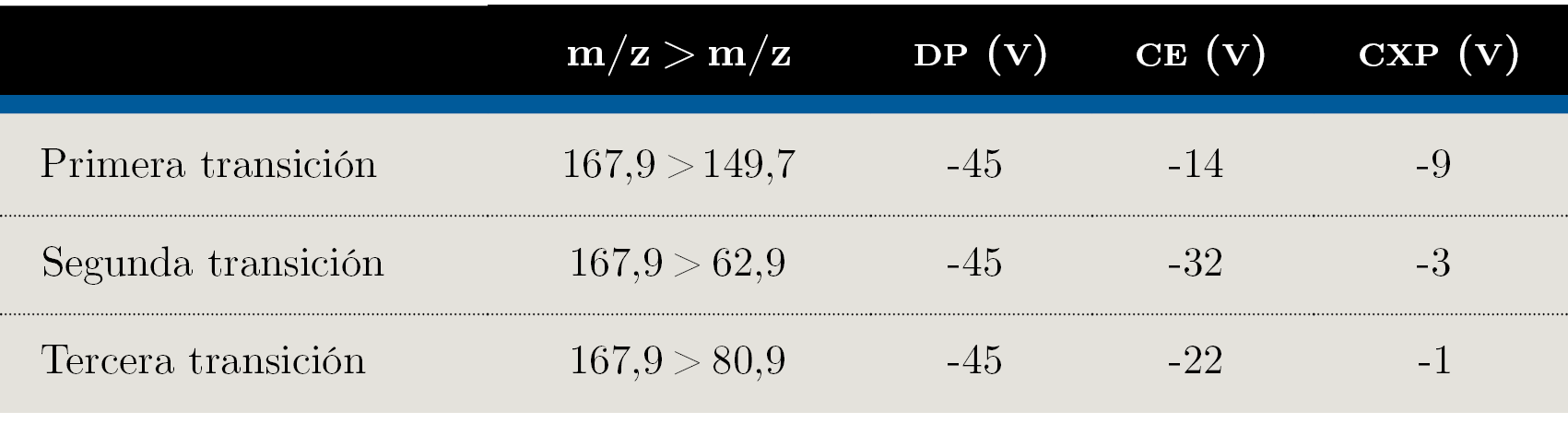
Para el análisis por LC–MS/MS se utilizó un cromatógrafo de líquidos Agilent 1200 (Agilent Technologies, Palo alto, CA, USA) acoplado a un sistema 4000 QTRAP LC– MS/MS de SCIEX (Framingham, Massachusetts, USA) en modo Scheduled MRM™. La separación cromatográfica se llevó a cabo dentro del horno a 40 °C utilizando una columna Dionex IonPac AS 11 (2 × 250 mm) equipada con una precolumna Dionex IonPac AG 11 (2 × 50 mm), ambas de Thermo Scientific. Se empleó un gradiente con una fase móvil compuesta por: A: agua; B: ácido cítrico 1 mM en agua ajustado a pH 11 con DMA, a un flujo constante de 300 μL min-1. El volumen de inyección fue 20 μL. La detección en MS/MS se realizó en modo monitoreo de reacciones múltiples (Multiple Reaction Monitoring, MRM) usando una interfase de ionización por electrospray (ESI) en modo de ionización negativa. El voltaje de ionización fue -4500 V, el gas de nebulización fue nitrógeno a 50 psi, y el curtain gas, nitrógeno a 20 psi. La evaporación por solvente en la fuente fue asistida por un gas para secado (nitrógeno a 50 psi calentado a 500 °C) y el gas de colisión también fue nitrógeno. Se realizaron experimentos utilizando una solución del analito a 0,1 mg L-1 para determinar las transiciones MRM óptimas, potencial de declustering (DP) y energías de colisión (CE). Para esto se utilizó una bomba de jeringa a flujo constante para la inyección directa al equipo de MS/MS de la solución de estándar. Los parámetros del MS/MS utilizados en este estudio están listados en la Tabla 1.
Preparación de muestras
Se ajustó la metodología de análisis denominada QuPPe. En un tubo de centrífuga se pesaron (5,00 ± 0,01) g miel, se agregaron 9 mL de H2O y se homogenizó utilizando un vortex. Luego, se adicionaron 10 mL de metanol acidificado con ácido fórmico al 1% y se agitó vigorosamente de forma manual y durante 1 minuto. El tubo se centrifugó 5 min a 4000 rpm. Finalmente, una alícuota de extracto se filtró a un vial de 2 mL con inserto de plástico para posteriormente inyectar en LC–MS/MS.
Resultados y discusión
Optimización de transiciones
Para comenzar a trabajar fue necesario ajustar las condiciones de operación del instrumento utilizado para la determinación de glifosato, en este caso un espectrómetro de masas en tándem de tipo triple cuadrupolo, operando en el modo de adquisición monitoreo de reacciones múltiples (Multiple Reaction Monitoring, MRM).
El tipo de ionización utilizado fue electrospray (ESI) en modo negativo, es decir, que en este caso se da la remoción de un protón de la molécula de glifosato, la cual queda cargada negativamente: pasa de un peso molecular de 169 (Figura 1) a tener, luego de la ionización, una relación masa/carga (m/z) = 168, con z = 1.
Los parámetros instrumentales optimizados para las transiciones de glifosato se muestran en la Tabla 1.
Tabla 1. Parámetros instrumentales optimizados en el espectrómetro de masas. DP: Declustering Potential; CE: Collision Energy; CXP: Cell Exit Potential.
Optimización de condiciones cromatográficas
El glifosato como tal es un compuesto compatible con cromatografía líquida. Para el análisis de los pesticidas muy polares se describen en el documento de EURL–SRM varias condiciones de LC–MS/MS. En particular para glifosato se enumeran tres condiciones posibles que han mostrado buenos resultados para el análisis de este herbicida: métodos 1.1, 1.2 y 1.3. En la Tabla 2 se indican sus principales características. Para adecuar la metodología conforme al instrumental y materiales con los que se contaba en el laboratorio fue necesario ajustar las condiciones cromatográficas. De acuerdo a esto, se trabajó con el método 1.1 “Glyphosate & Co. AS 11” para el cual se utilizó una columna de intercambio aniónico Dionex IonPac AS 11 (2 × 250 mm) conectada a una precolumna Dionex IonPac AG 11 (2 × 50 mm), ambas mantenidas en un horno a 40 °C. El volumen de inyección fue de 20 µL.
Tabla 2. Condiciones de LC–MS/MS utilizadas para el análisis de glifosato. Modificado de Anastassiades et al. (2017).

Las fases móviles empleadas fueron agua y una solución de ácido cítrico 1 mM en agua, ajustada a pH 11 con dimetilamina (DMA). En la Tabla 3 se muestra el gradiente utilizado, con un tiempo total de corrida de 23 minutos.
Tabla 3. Gradiente utilizado en la corrida cromatográfica. A = agua. B = ácido cítrico 1 mM en agua, ajustado a pH 11 con dimetilamina (DMA).

Método de extracción
El método utilizado para la extracción de glifosato de la miel fue el descrito en el documento de EURL – SRM para el análisis de varios pesticidas muy polares (denominado método QuPPe) en alimentos, dentro de los cuales se encuentra la miel como producto procesado. Esta metodología consiste en una extracción de los residuos de la muestra con metanol acidificado, previo ajuste del contenido de agua en caso de ser necesario. La cantidad de agua esperada en el extracto es 10 g, incluyendo el contenido natural de agua de la muestra y lo que deba agregarse para alcanzar esa cantidad; para el caso de la miel, fue necesaria la adición de 9 mL de agua a los 5 g de muestra.
Validación de la metodología
Para realizar la validación del método, se siguió el documento SANTE/11813/2017 de la Unión Europea, en el cual se establece una guía sobre procedimientos analíticos de control de calidad y validación de métodos para el análisis de residuos de pesticidas en alimentos y piensos (European Commission, Directorate General for Health and Food Safety, 2017).
Se llevaron a cabo ensayos de recuperación a tres niveles de concentración: 0,025 mg kg-1, 0,050 mg kg-1 y 0,075 mg kg-1. Los ensayos se hicieron fortificando una muestra de miel blanco, es decir, que no contuviera glifosato, con una cantidad conocida del pesticida. Para cada nivel de concentración se hicieron cinco réplicas, a partir de las cuales se calculó el porcentaje de recuperación (Ecuación 1) para evaluar la veracidad del método y la desviación estándar relativa (RSD, por sus siglas en inglés) (Ecuación 2), con el fin de evaluar su precisión.
$${Rec\thinspace(\%)=\frac{Concentraci\acute{o}n_{experimental}}{Concentraci\acute{o}n_{te\acute{o}rica}}\times100}$$ (1)
$${RSD\thinspace(\%)=\frac{\sigma}{Rec_{promedio}}}\times100$$ (2)
El documento SANTE (European Commission, Directorate General for Health and Food Safety, 2017) establece como parámetros aceptables que el porcentaje de recuperación se encuentre entre 70% y 120%, con una RSD menor a 20%. Como límite de cuantificación del método se considera el nivel de concentración más bajo evaluado en el proceso de validación y que cumpla con los criterios descritos anteriormente. Como se puede observar en la Tabla 4, a los tres niveles ensayados se cumplen las condiciones. Por lo tanto, el límite de cuantificación del método fue establecido en 0,025 mg kg-1. En la Figura 2 se ejemplifican los cromatogramas obtenidos al nivel del límite de cuantificación.
Tabla 4. Porcentajes de recuperación y desviaciones estándar relativas (RSD) obtenidos a los tres niveles de concentración ensayados.
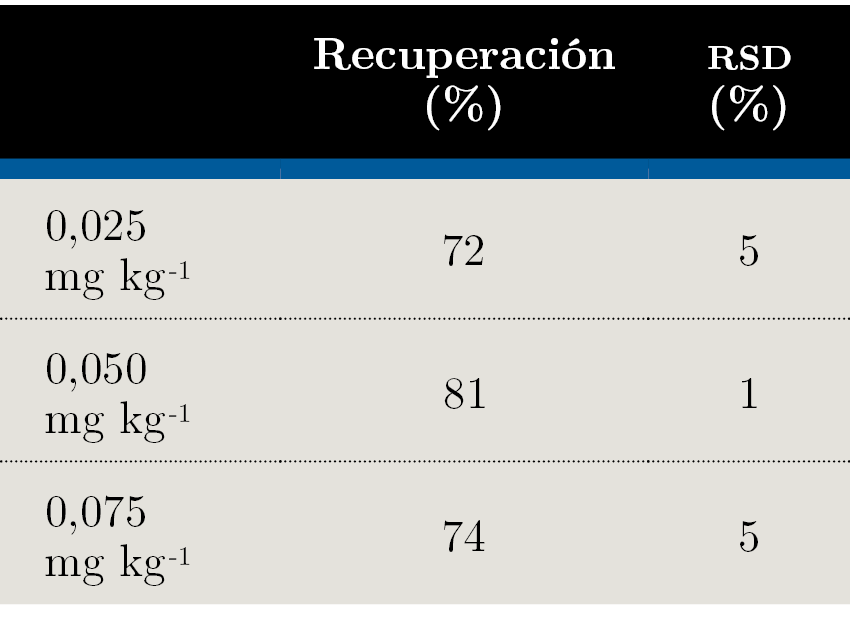
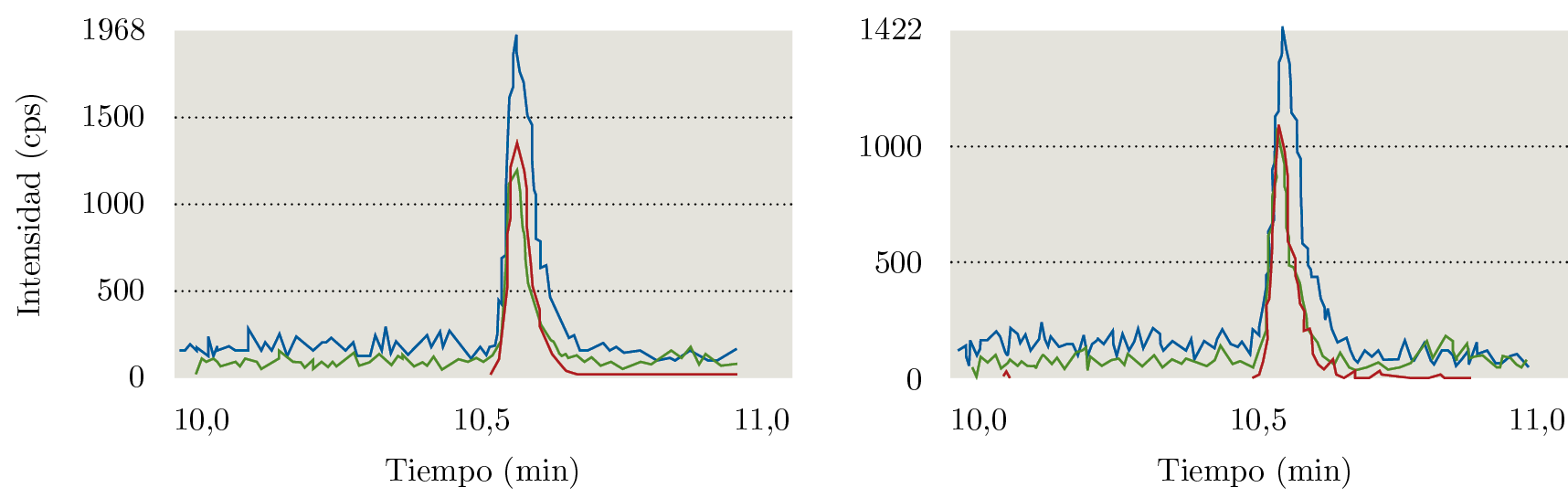
Figura 2. Cromatogramas de iones extraídos de glifosato en: a) punto de calibración en matriz a 0,025 mg kg-1 y b) recuperación a 0,025 mg kg-1.
Por otro lado, se evaluó la linealidad de la respuesta instrumental con respecto a la concentración del analito. El estudio de la linealidad se llevó a cabo a partir de la construcción de curvas de calibración tanto en solvente como en matriz (Figura 3). La linealidad fue evaluada de acuerdo al criterio de ± 20% de desviación de la concentración calculada mediante la curva de calibración con respecto a la teórica evaluada en cada punto (Ecuación 3). Además, se realizó una inspección visual del gráfico y se calculó el coeficiente de determinación (R2). Se observó un comportamiento lineal del glifosato tanto en solvente como en matriz entre 0,010 y 0,500 mg kg-1, ya que se obtuvo un coeficiente de determinación apropiado y para todos los puntos se tuvo una desviación menor a ± 20%, tal como establece el documento SANTE (European Commission, Directorate General for Health and Food Safety, 2017).
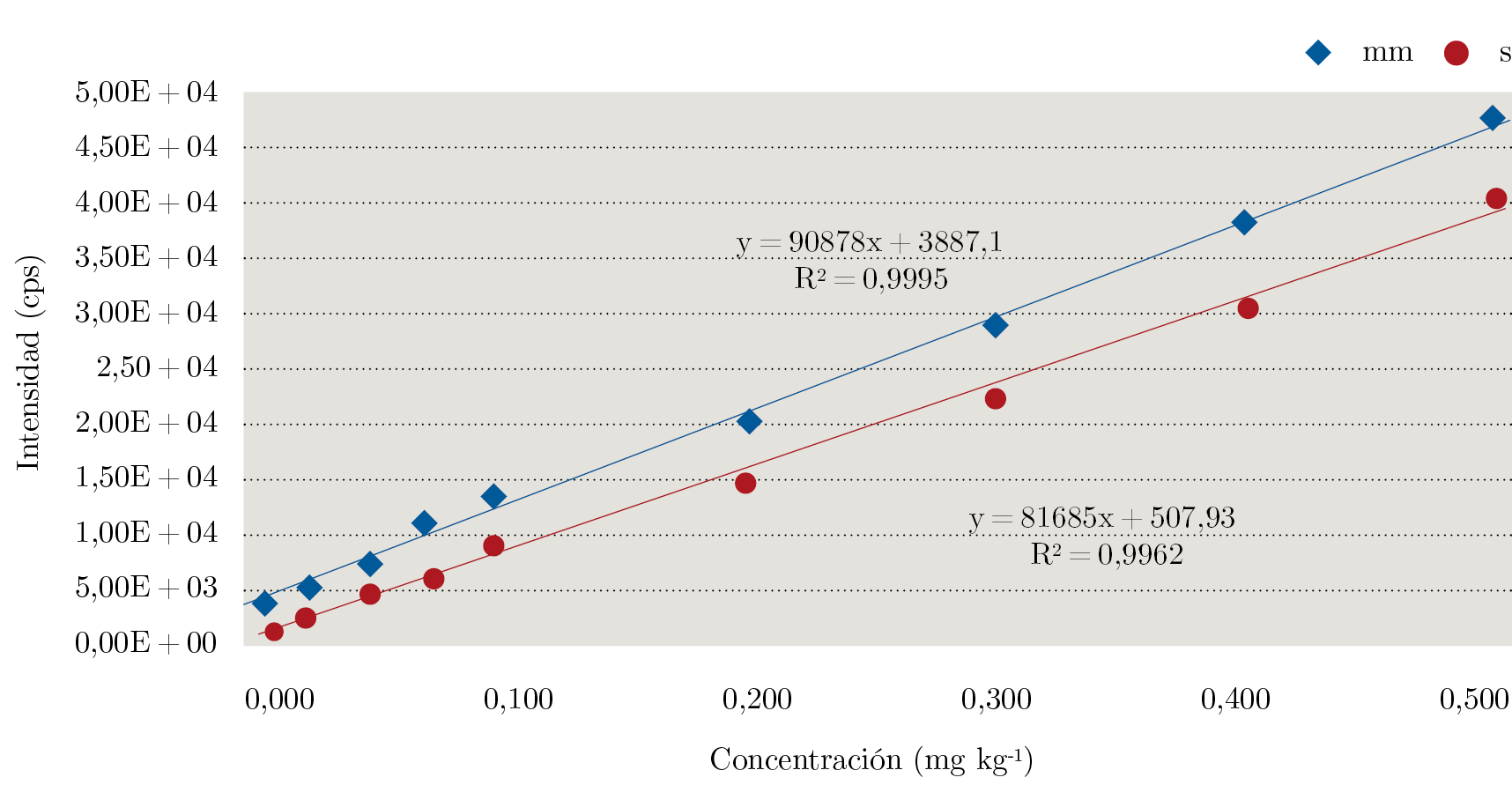
Figura 3. Curvas de calibración en matriz (mm) y en solvente (s) para glifosato.
$${Desviación\:de\:la\:concentración\:calculada\:(\%)=100\times\frac{C_{calculada}-C_{te\acute{o}rica}}{C_{te\acute{o}rica}}}$$ (3)
La curva de calibración en matriz (en inglés Matrix – matched calibration) se construye agregando cantidades crecientes conocidas de pesticida a varias alícuotas de extracto blanco, y es útil para cuantificar correctamente cuando se trabaja con matrices complejas en las cuales por lo general se observa efecto matriz, es decir, cuando existe una supresión o incremento de la señal instrumental por la presencia de otros componentes de la matriz que coeluyen con el analito. El efecto matriz para glifosato en el extracto de miel fue evaluado y calculado como el porcentaje de la diferencia entre la pendiente de la curva en matriz con respecto a la pendiente de la curva en solvente (Ecuación 4). Se observó un incremento en la señal, con un porcentaje de efecto matriz del 12%, lo cual no es lo más común, ya que por lo general, al trabajar con cromatografía líquida acoplada a un espectrómetro de masas con una fuente de ionización por electrospray, se observa supresión de la señal. Esto se produce porque los componentes de la matriz que coeluyen con los analitos de interés “compiten” por la carga, disminuyendo la eficiencia de la ionización de estos últimos. Es de destacar la importancia de cuantificar utilizando la curva de calibración preparada en matriz, ya que de prepararla en solvente se podría estar cuantificando de manera errónea un resultado positivo.
$${EM\thinspace(\%)=\frac{pendiente_{mm}-pendiente_s}{pendiente_s}}\times100$$ (4)
Análisis de muestras reales
Para evaluar el desempeño del método, se analizaron 15 muestras reales de mieles uruguayas, en 13 de las cuales se encontraron residuos de glifosato. De las 13 muestras que tuvieron un resultado positivo, el 70% contenía una concentración por encima del límite de cuantificación del método (0,025 mg kg-1), en un rango que varió hasta 0,500 mg kg-1.
Conclusiones
El método ajustado es sencillo y podrá ser empleado para el control de calidad e inocuidad de la miel en lo que a residuos de glifosato respecta, ya que el LMR de glifosato para la miel según la Unión Europea está establecido como 0,050 mg kg-1.
Además, esta herramienta analítica validada bajo las condiciones del laboratorio del Grupo de Análisis de Compuestos Traza (GACT) de la Universidad de la República es un insumo que contribuye a asegurar la inocuidad del alimento a ser consumido por la población y a la sustentabilidad de la cadena apícola uruguaya, que tiene participación en el PBI del país y que representó el 0,3% del total de las exportaciones de origen agropecuario en el año 2017 (DIEA, 2018).
La metodología ajustada y validada es una buena herramienta, porque además contribuirá a generar insumos para utilizar la colmena como un indicador de sustentabilidad de agroecosistemas.
Reconocimientos
Los autores agradecen a RALACA–IAEA por el suministro del estándar de glifosato, a INIA en el marco del proyecto FPTA–320 y al Quinto Congreso Uruguayo de Química Analítica (CUQA).