Autores
- Taborda, Belén (1); Telles, Inés (1); Pareja, Lucía (1); Niell, Silvina (2); Jesús, Florencia (2); López, Andrés (1)
- Departamento de Química del Litoral, Cenur Litoral Norte, UdelaR, Paysandú, Uruguay
- Departamento de Química del Litoral, PDU Abordaje Holístico al impacto de los agroquímicos, Cenur Litoral Norte, UdelaR, Paysandú, Uruguay
En este trabajo se compararon tres diferentes métodos de extracción de lípidos en semillas de quinoa utilizando: maceración dinámica con hexano, maceración con hexano asistida por ultrasonido y extracción continua en Soxhlet con gradiente de polaridad (hexano, acetato de etilo, metanol). Para cada una de las metodologías evaluadas se determinó el rendimiento y posteriormente la derivatización para generar los ésteres metílicos de los ácidos grasos (FAMEs) correspondientes. Todas las fracciones obtenidas (extractos y FAMEs) fueron evaluadas mediante cromatografía en capa fina. Se observó que los métodos de extracción que utilizan hexano y acetato de etilo como solvente presentaron manchas de similar concentración y factor de retención. Mientras que el extracto obtenido con metanol presentó manchas de similar factor de retención que las fracciones de hexano pero con menor intensidad, también mostró otras manchas muy polares que permanecieron en la línea de siembra. Las diferentes fracciones en hexano transesterificadas se analizaron mediante cromatografía gaseosa con detector de ionización de llama, con detector de masas y mediante resonancia magnética nuclear. Por medio de GC-FID se identificaron los siguientes ácidos grasos: C 14:0; C16:0; C18:0; C18:1; C18:2; C18:3 y C20:1. El análisis mediante GC-MS permitió la confirmación mediante biblioteca de C16:0; C18:0; C18:1; C18:2.
Palabras clave: quinoa, GC-MS, GC-FID, Soxhlet, maceración dinámica, maceración asistida mediante ultrasonido.
Introducción
La quinoa (Chenopodium quinoa Willd) es un pseudocereal nativo de la región Andina, de hoja ancha anual de semillas blancas, rojas y negras. El término pseudocereal se debe a que, si bien comparte la mayoría de las características de crecimiento de los cereales, es una planta dicotiledónea, a diferencia de los cereales, que son monocotiledóneas (Przybylski, et al., 1994). Las semillas son pequeñas, aplanadas y de forma ovalada, de 1,5 mm de diámetro; 350 semillas pesan aproximadamente un gramo. Es un cultivo de bajos requerimientos, entre ellos, poca lluvia, elevada altitud, bajas temperaturas y suelos alcalinos arenosos (Marmouzi, et al., 2015).
Existe una alta demanda mundial dado su alto valor nutricional, particularmente por el contenido de proteínas y de aminoácidos, además de lípidos beneficiosos (Prego, et al., 1998) La calidad de las proteínas de quinoa está reportada como equivalente a las de la leche. Un inconveniente a destacar es la presencia de saponinas en el pericarpio, por lo cual se le atribuye su sabor amargo y la formación de espuma en agua. Por esta razón antes de su consumo se recomienda realizar uno o varios enjuagues hasta la desaparición de espuma.
Los principales ácidos grasos reportados por bibliografía son: ácido mirístico, ácido palmítico, ácido esteárico, ácido docosanoico, ácido miristoleico, ácido palmitoleico, ácido oleico, ácido linoleico y ácido linolénico. La presencia de ácido linoleico y ácido linolénico hacen a los aceites susceptibles a la rancidez oxidativa, lo cual no ocurriría en el caso del aceite de semilla de quinoa, debido a su contenido de antioxidantes (Rubio, 2005).
El alto consumo de quinoa y de otros pseudocereales en Uruguay motiva el interés de determinar la composición de lípidos comparando diferentes metodologías para establecer cuál es la más adecuada para la extracción de los lípidos de quinoa y realizar su caracterización mediante cromatografía gaseosa acoplada a diferentes detectores y resonancia magnética nuclear.
Materiales y Métodos
Material vegetal
Se utilizaron semillas de quinoa comerciales (La Abundancia, obtenidas de un supermercado local en la ciudad de Paysandú), que se molieron en un molino de corte (IKA-M20) hasta obtener una harina. La harina de quinoa se guardó en un ambiente seco hasta el momento del análisis (Fanalli, et al., 2015).
Extracción
Maceración dinámica
En un vaso de Bohemia de 250 mL se pesaron 50 g de harina de quinoa y se agregaron 100 mL de hexano, se agitó con agitador magnético por 30 min a temperatura ambiente. Se dejó decantar, se tomó la fase orgánica y se adicionaron nuevamente 100 mL de hexano sobre el marco vegetal, agitando por 30 min. Se filtró, se reunieron ambos extractos y se llevó a sequedad mediante destilación a presión reducida a 40 °C (Fanalli, et al., 2015).
Maceración asistida con ultrasonido
En un Erlenmeyer de 250 mL se pesaron 25 g de harina de quinoa, se agregaron 100 mL de hexano y se llevó al ultrasonido por 30 min a temperatura ambiente. Se filtró directamente a un Erlenmeyer con papel de filtro en embudo por gravedad y el extracto se llevó a sequedad mediante destilación a presión reducida a 40 °C (Pellegrini, 2018).
Extracción continua en Soxhlet
Se pesaron 50 g de harina de quinoa en un dedo de papel de filtro que se colocó en el Soxhlet, y se dejaron en reflujo con 360 mL de cada solvente durante 30 min. Se realizó un gradiente creciente de polaridad utilizando como solventes hexano, acetato de etilo y metanol, en ese orden, para la extracción de distintos compuestos del marco vegetal. Los extractos orgánicos se llevaron por separado a sequedad por destilación a presión reducida a 40 °C (Marmouzi, et al., 2015).
Transesterificación
Se realizó una derivatización a cada uno de los extractos obtenidos de los tres métodos de extracción evaluados (maceración dinámica, extracción asistida mediante ultrasonido y Soxhlet con los tres solventes seleccionados) para su posterior análisis. El procedimiento realizado para la preparación de los ésteres metílicos consistió en tomar una alícuota de extracto correspondiente a 50 mg en un tubo de tapa a rosca, luego se agregaron 2 mL de solución metanólica de KOH 0,5 N y se agitó por 10 min. A continuación, se calentó a 60 °C por 10 min y se dejó enfriar a temperatura ambiente. Se agregaron 2 mL de solución metanólica de HCl 1 N y se calentó en baño de agua por 5 min. Se dejó enfriar y se extrajo con 2 mL de hexano. Se transfirió la fase orgánica a un vial secando con Na2SO4 anhidro y se filtró con filtro de 0,45 µm, según método tomado y modificado de Darnoko et al. (2000).
Detección por cromatografía en capa fina
Se utilizaron placas de fase normal de sílica gel como fase estacionaria (TLC Sílica gel 60 F254, 20 × 20 cm) y una mezcla hexano: acetato de etilo: ácido fórmico (90:10:2) como fase móvil (Huang, et al., 2015). El revelador utilizado para la evaluación de todos los extractos y sus transesterificaciones fue revelador universal de CuSO4 (CAMAG, 2018).
Análisis por GC-FID
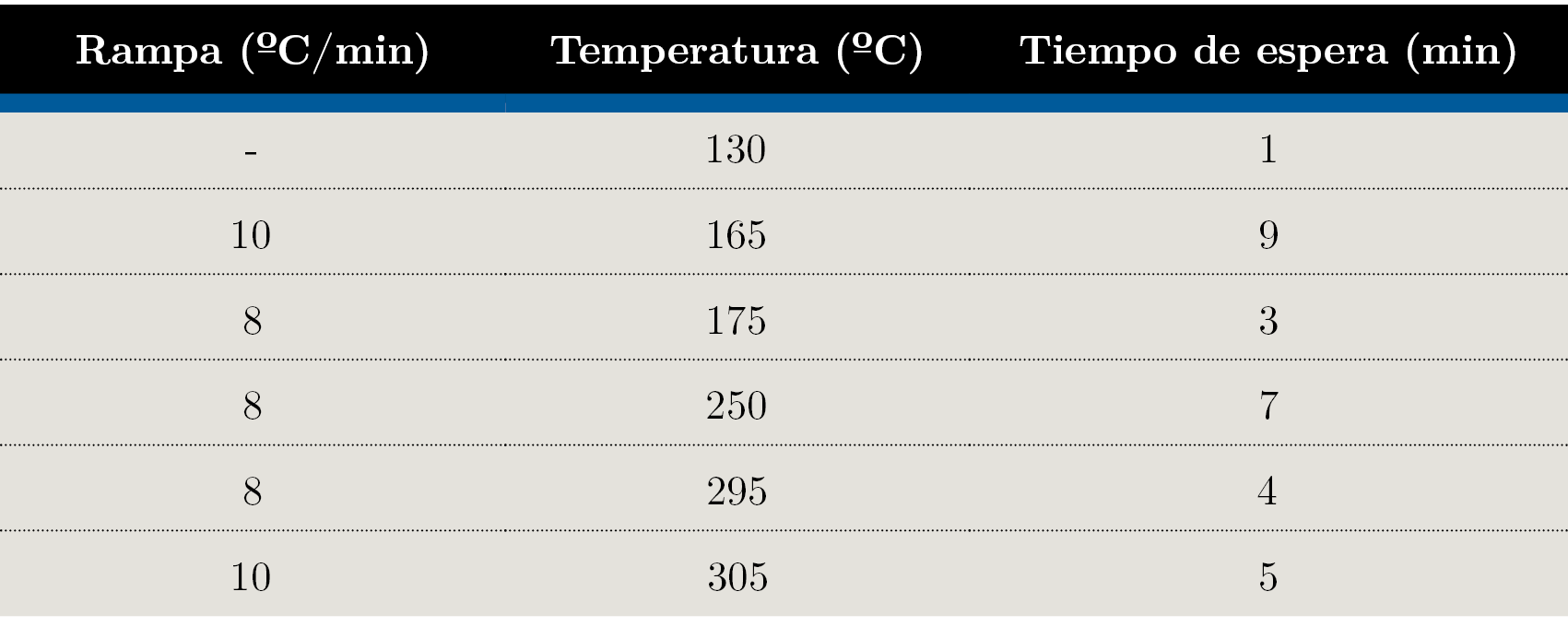
El análisis de los extractos de Soxhlet transesterificados fue realizado en un cromatógrafo de gases equipado con un inyector manual modelo GC2010 Plus (Shimazdu) y una columna TR-FAME (Thermo 120 m × 0,25 mm × 0,25 µm). Las condiciones experimentales fueron: inyección Split, relación de Split 1:50; temperatura del inyector 280 °C; temperatura del detector 250 °C; volumen de inyección 1 μL; gas carrier Nitrógeno. El programa de temperatura se muestra en la Tabla 1, con un tiempo total de corrida de 99 min.
Tabla 1. Programa de temperatura utilizado para análisis GC-FID.

Análisis por GC-MS
El análisis se realizó en un equipo GC Shimazdu 2010 equipado con inyector automático Shimazdu AOC-20i y una columna TR-5MS (Thermo 30 m × 0,25 mm × 0,25 µm). Las condiciones experimentales fueron: temperatura del inyector 280 °C; temperatura de la fuente de iones 230 °C; volumen de inyección 1 μL; inyector split, relación de Split 1:10; gas carrier Helio; 1.0 mL/min. El programa de temperatura se muestra en la Tabla 2, con un tiempo total de corrida de 50,15 min. La ionización fue por impacto electrónico 70 eV y el rango de masas 50-550 m/z.
Tabla 2. Programa de temperatura utilizado para análisis GC-MS.
Análisis por RMN
El espectro de RMN de protón se adquirió a 25 °C en un espectrómetro Bruker Avance III 500 MHz equipado con una sonda inversa de triple resonancia (TXI 1H-13C/15N) de 5 mm. Se disolvieron 10 mg de cada fracción del Soxhlet en 600 µL de CDCl3, con 0,03 % de tetrametilsilano (TMS) como estándar interno. Se realizaron 64 scaners con tiempo de relajación de 10 mseg.
Resultados y Discusión
Obtención de fracción lipídica
El rendimiento de cada una de las metodologías se evaluó por duplicado y se calcularon a partir de la masa obtenida luego de la extracción y en función de la masa inicial utilizada en cada uno de los métodos seleccionados y fueron expresados en porcentaje. Los rendimientos fueron 1,6 y 2,1% para maceración dinámica con hexano y maceración asistida con ultrasonido. Los rendimientos obtenidos para las tres fracciones obtenidas mediante Soxhlet fueron: fracción hexano 1,9 %, fracción acetato de etilo 2,0 %, fracción metanol 2,1 %. El mayor rendimiento obtenido con metanol puede deberse a que, además de extraer el remanente de lípidos luego de la extracción exhaustiva con hexano y acetato de etilo, este solvente por su polaridad extrae componentes polares que no son extraídos con los demás solventes.
Detección por TLC
Con el fin de comparar las diferentes metodologías de extracción, se sembraron las fracciones en una placa de sílica gel de 10 cm × 12 cm, utilizando como fase móvil una mezcla hexano-acetato de etilo-ácido fórmico (90:10:2). Las fracciones sembradas fueron: extracto de maceración dinámica con hexano; extracto de maceración asistida con ultrasonido en hexano; extractos de Soxhlet con hexano, acetato de etilo y metanol; estándar de sitosterol; estándar de ácido mirístico, y los extractos transesterificados de maceración dinámica hexano, Soxhlet con hexano y Soxhlet con acetato de etilo. En la TLC se pudieron observar las manchas características de los ésteres metílicos de ácidos grasos demostrando que se llevó a cabo una correcta transesterificación. En el punto de siembra de la fracción metanólica del Soxhlet se puede confirmar la presencia de compuestos muy polares debido a su retención sobre la sílica. Las manchas del extracto de Soxhlet en acetato de etilo fueron similares a las de hexano, pero de menor intensidad. Se observó también que las distintas extracciones que utilizan como solvente hexano presentan las mismas manchas y con la misma relación de frente, aproximadamente de 0,8 cm.
Análisis de ésteres metílicos de ácidos grasos por GC-FID
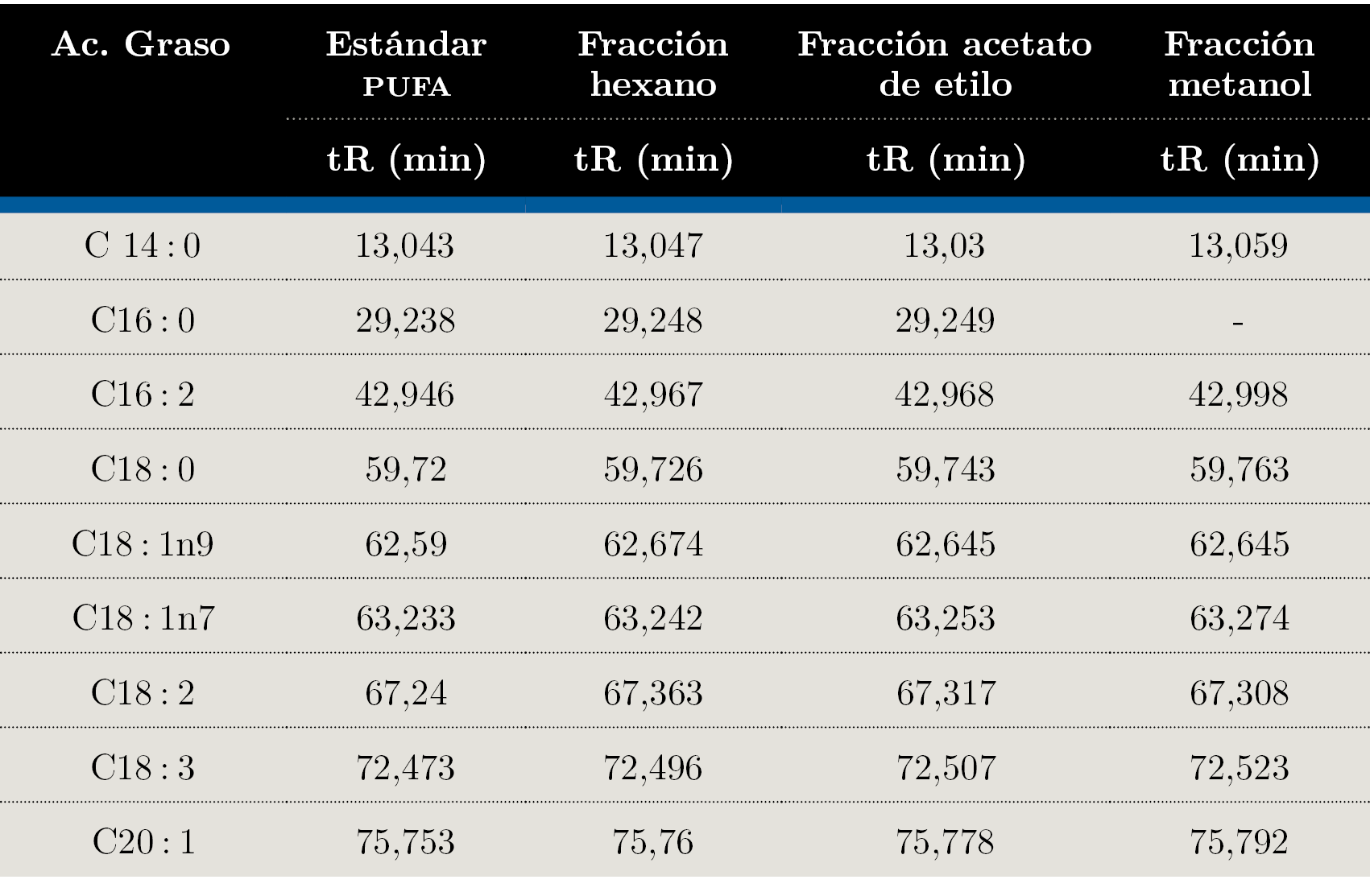
Debido a que los tres extractos obtenidos mediante Soxhlet fueron los que presentaron distintos comportamientos en TLC se seleccionaron para realizar el análisis mediante GC-FID. En la Figura 1 se muestra el orden elución para el estándar (PUFA 3: ácidos grasos poli-insaturados) obtenidos con la columna analítica seleccionada (Figura 1) y el orden de elución de los componentes en muestra de la fracción de hexano (Figura 2). La identificación de los AG se realizó por medio de la comparación de los tiempos de retención obtenidos para cada una de las muestras con el análisis del estándar PUFA 3: AG poli-insaturados obtenido en las mismas condiciones de análisis (Tabla 3).
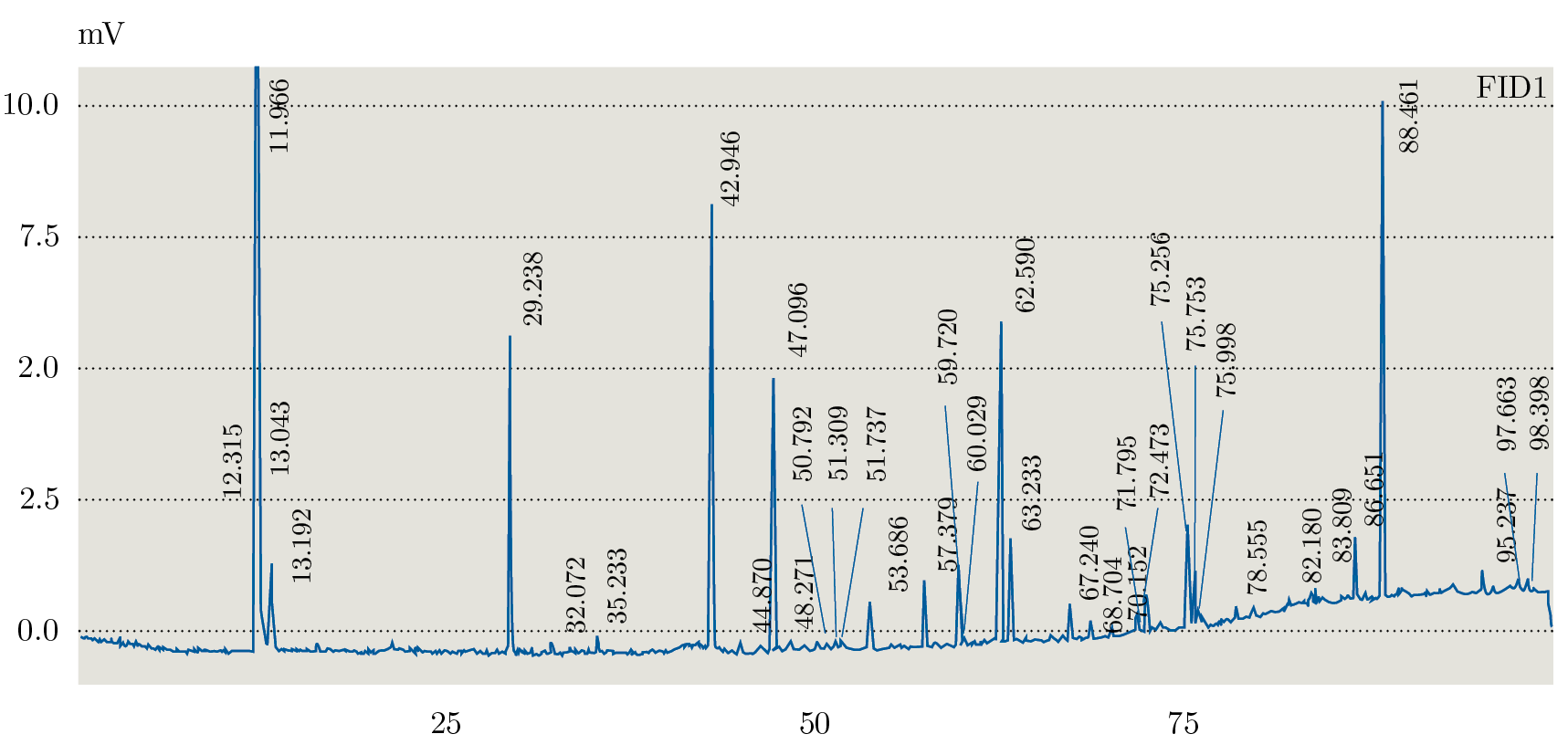
Figura 1. Cromatograma GC-FID del estándar PUFA.
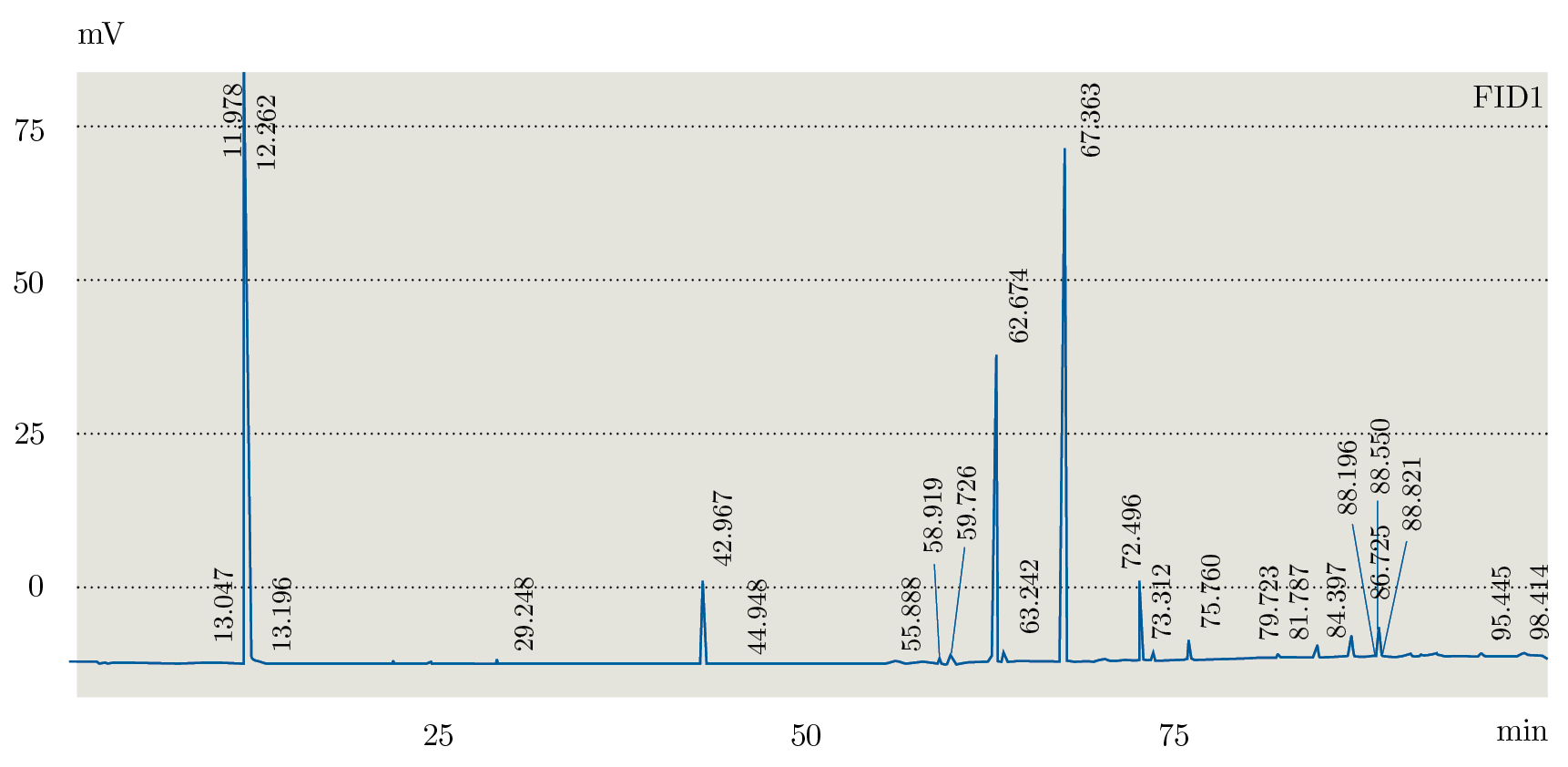
Figura 2. Cromatograma GC-FID de extracción en Soxhlet fracción hexano.
A partir de los análisis realizados mediante GC-FID se identificaron los siguientes ácidos grasos: C14: 0; C16: 0; C16: 2; C18: 0; C18: 1n9; C18: 1n7; C18: 2; C18: 3; C20: 1. Para la fracción de acetato de etilo se identificaron C14: 0; C16: 0; C16: 2; C18: 0; C18: 1n9; C18: 1n7; C18: 2; C18: 3; C20: 1. La fracción de metanol presentó C14:0; C16:2; C18:0; C18:1n9; C18:1n7; C18:2; C18:3; C20:1. No se observó el C16:0 en esta fracción. Como era de esperar, los extractos de hexano y acetato de etilo presentaron mayor cantidad de AG que la fracción de metanol, verificando los resultados obtenidos mediante TLC.
Tabla 3. Tiempos de retención en minutos para cada pico identificado.
Análisis de ésteres metílicos de ácidos grasos por GC-MS
En los cromatogramas obtenidos para cada una de las fracciones se observaron los mismos picos correspondientes a los ácidos grasos: C16: 0; C18: 0; C18: 1; C18:2. Estos coincidieron en tiempo de retención con el estándar PUFA. A su vez se compararon con biblioteca y se confirmó su identificación (Figura 3).
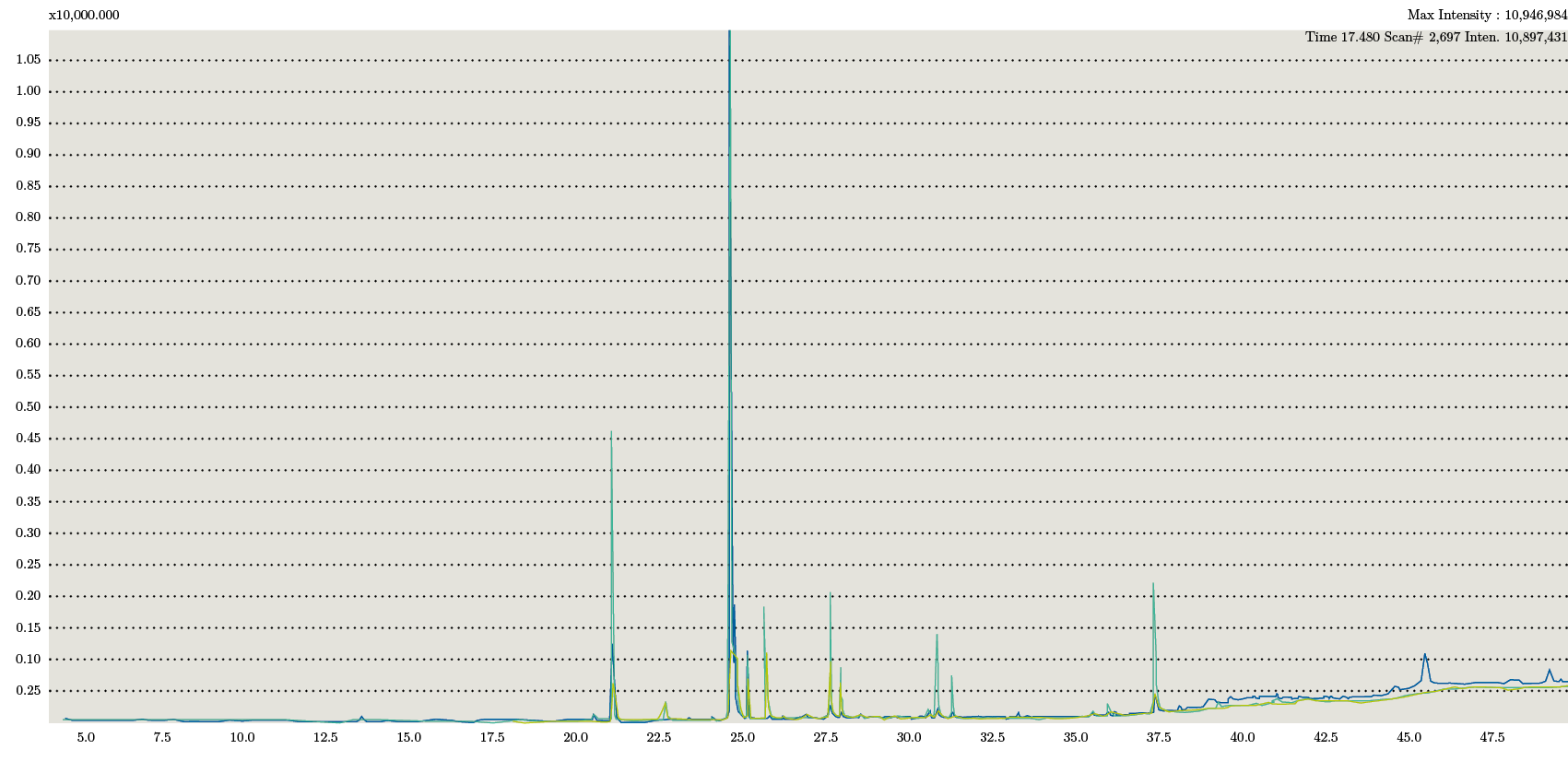
Figura 3. Superposición de cromatogramas de los tres extractos correspondientes a la extracción mediante Soxhlet.
Análisis por RMN
Se realizaron los espectros para todas las fracciones obtenidas en la extracción continua en Soxhlet. En la Figura 4 se observa el espectro obtenido correspondiente a la fracción de hexano. Se pueden observar los picos característicos del perfil lipídico, con los diferentes tipos de protones asignados. Los resultados confirman la presencia de TAG (triacilgliceroles), ácidos grasos saturados e insaturados. También se observa que en la fracción metanólica se coextraen junto con los lípidos otros compuestos que dificultan la integración y resolución de los picos para su caracterización.
Estos espectros permiten caracterizar la matriz según los siguientes parámetros: el índice de olefinas (IO: moles olefinas por mol de AG); ácidos grasos insaturados (IFA: moles AG que presentan al menos una insaturación por mol de ácido graso); MUFA (moles de olefinas totales menos los moles de olefinas conjugadas por mol de AG) y PUFA (protones bisalílicos por mol de AG), como se detalla en la Tabla 4.
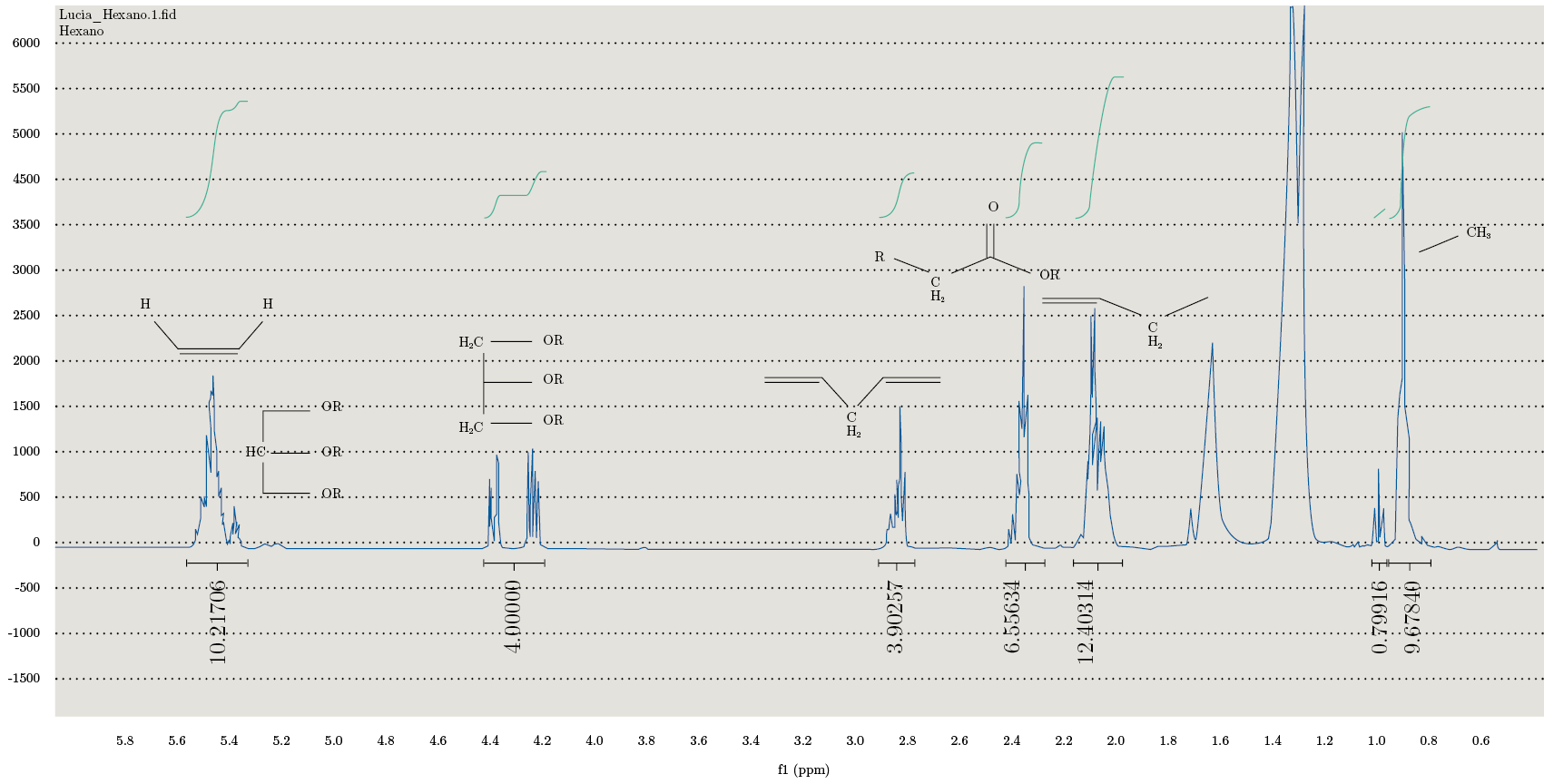
Figura 4. Espectro de RMN-1H de la fracción correspondiente a la extracción obtenida mediante Soxhlet con hexano.
Tabla 4. Datos de integración del espectro de RMN e índices obtenidos para para las fracciones correspondientes a la extracción continua en Soxhlet con hexano y acetato de etilo.

La comparación de las metodologías seleccionadas muestra que los rendimientos de extracción son similares. Además, el análisis mediante TLC confirma que con todas las metodologías se obtienen los mismos compuestos mayoritarios, a excepción, como era de esperar, de la fracción obtenida mediante la extracción continua a partir de Soxhlet en metanol.
El estudio de las fracciones de la extracción por Soxhlet mediante GC-FID utilizando una columna cromatográfica específica para ésteres metílicos de ácidos grasos permite identificar los picos mayoritarios, que a su vez se corresponden con los reportados por bibliografía.
Los principales ácidos grasos extraídos se confirmaron mediante análisis por GC-MS y RMN, y se calcularon algunos parámetros de interés: IO, IFA, MUFA y PUFA.