Autores
- Gérez García, Natalia (1); Bertón Passarino, Analía (1); Gil Borba, Guillermo (1); Cesio, María Verónica (1, 2); Heinzen, Horacio (1, 2)
- Grupo de Análisis de Compuestos Traza (GACT), DQO, Facultad de Química, UdelaR, Montevideo, Uruguay.
- Grupo de Análisis de Compuestos Traza (GACT), DQL, CenUR Litoral Norte, UdelaR, Paysandú, Uruguay.
Dentro de los ectoparasiticidas utilizados en producciones ganaderas, en las que los animales y los procesos industriales generan productos para consumo humano, como la lechería, existen diferentes combinaciones de pesticidas y acaricidas. Si no se administran adecuadamente y no se respetan los compases de espera, se pueden encontrar sus residuos en los alimentos, por ejemplo, en los productos lácteos. Este fenómeno representa un potencial peligro para los consumidores, por lo que es necesario generar herramientas que permitan determinar cómo afectan las diferentes formas de uso en las concentraciones de pesticidas a los alimentos y los intervalos que aseguren su inocuidad al consumirlos.
El objetivo de este trabajo fue ajustar y validar una metodología multirresiduo de análisis en leche vacuna cruda para al determinacion de los ectoparasticidas más empleados en ganadería utilizando GC-QqQ-MS, método elegido por su selectividad y especifidad. La preparación de muestra se basó en la metodología oficial para el análisis de residuos de pesticidas en alimentos en los EE.UU.: QuEChERS, AOAC 2007.01 (AOAC International, 2016). Se validó la metodología según el documento SANTE vigente, evaluando veracidad, precisión, linealidad, efecto matriz y límites de cuantificación (European Commission. Directorate General for Health and Food Safety, 2017).
Esta metodología es una herramienta útil para determinar curvas de decaimiento de los compuestos aplicados de manera de asegurar la calidad e inocuidad de la leche vacuna cruda.
Palabras clave: Pour-on, residuos de pesticidas en leche cruda, GC-MS/MS, validación de metodología.
Introducción
La eliminación de xenobióticos en mamíferos se realiza a través de los fluídos que el organismo excreta. Si bien las vías usuales de eliminación son la orina y las heces, otros fluídos que se producen en gran cantidad, como la leche en períodos de amamantamiento de la cría, se tornan relevantes. En particular, la leche vacuna es un alimento producido a escala industrial y altamente consumido por la población. Para mantener la productividad de los animales es crítico preservar su bienestar y salud. En ese marco, el uso de medicamentos veterinarios es una práctica corriente.
En particular, para evitar los problemas de estrés que genera la mosca de los cuernos, es común el uso de ectoparasiticidas tipo pour-on. Estos contienen insecticidas que luego de ser absorbidos se distribuyen sistémicamente y pueden alcanzar la leche de las vacas productoras. Este fenómeno obliga al establecimiento de períodos de espera en los que la leche obtenida no puede ser consumida porque contiene altos niveles de esos compuestos.
Debido a su contenido en grasas relativamente alto, la leche es un vehículo para la eliminación de contaminantes lipofílicos y ha sido empleada para monitorear la bioconcentración de contaminantes orgánicos persistentes, tales como los pesticidas organoclorados (González-Rodríguez, et al., 2005), ya que está comprobado que estos se acumulan en los tejidos adiposos y son excretados en asociación con la porción grasa de la leche (Trujillo-Parra, et al., 2003).
Otros pesticidas lipofílicos pero menos persistentes o estables, como por ejemplo los organofosforados, también han sido encontrados en leches de diversos orígenes, como la leche procesada de origen animal o leche materna sin procesar.
Si bien una de las vías de contaminación de la leche vacuna puede deberse al uso de drogas veterinarias, las razones para la contaminación pueden ser de diversa índole, como por ejemplo tratamientos higiénicos contra insectos en plantas de procesamiento de leches, consumo de productos alimenticios que contengan pesticidas o uso de raciones para animales que contienen materiales vegetales tratados durante la temporada de cultivo con pesticidas, entre otros. Debido a que los pesticidas son compuestos orgánicos peligrosos, la determinación de sus residuos en alimentos como la leche es importante por el riesgo que suponen para la salud humana. A tales efectos, las principales agencias reguladoras han fijado límites máximos de residuos (LMRs) que sirven para asegurar la inocuidad de su consumo (Codex Alimentarius, 2018). Dado que los principales consumidores de leche son los niños y bebés, los límites para estas franjas etarias son mucho más estrictos. Esto obliga a emplear métodos analíticos con una capacidad de detección hasta niveles de concentración mucho menores que los alcanzados por los métodos analíticos empleados habitualmente.
La determinación de residuos de pesticidas en leche cruda presenta problemas por tratarse de una matriz compleja con alto contenido graso y proteico que forma una emulsión O/W. Para su determinación en esta matriz compleja se han desarrollado diversos métodos (Souza, et al., 2016).
Los métodos reportados para determinar residuos de pesticidas en leche están basados en extracciones líquido-líquido, como el método de Luke, que emplea acetona, el método SweEt, que utiliza Acetato de Etilo como solvente de extracción, y el de QuEChERS (Quick, Easy, Cheap, Effective, Rugged y Safe), cuyo solvente extractor es acetonitrilo, así como otros métodos que involucran extracción en fase sólida, como el de Dispersión de la Matriz en Fase Sólida (MSPD). Desde hace 15 años, el análisis de residuos de pesticidas está dominado por los conceptos de miniaturización y economía en el uso de reactivos con el objetivo de cumplir con los parámetros de la química verde. Para acompasar las necesidades actuales se deben emplear técnicas de alta sensibilidad como la espectrometría de masas en tándem. Dentro de los métodos descritos, el de QuEChERS es el más empleado para el análisis multirresiduos de pesticidas en alimentos y productos agrarios, ya que permite obtener altas recuperaciones, resultados precisos, rapidez de tratamiento, poco uso de solvente y material de vidrio. Además, requiere poco espacio de laboratorio y bajo consumo de reactivos (Anasstasiades, et al., 2003). Asimismo, el proceso es robusto y confiable y consta de dos etapas: extracción y clean-up. La etapa de extracción utiliza acetonitrilo (MeCN) y sulfato de magnesio anhidro (MgSO4), lo cual mejora las recuperaciones al facilitar la partición de los pesticidas en la capa orgánica gracias a que retiene el agua. En el caso de la leche, la alta fuerza iónica que se genera ayuda a la precipitación de las proteínas. El método QuEChERS original no ajusta el pH de extracción (Anastassiades, et al., 2003), por lo que aquellos analitos lábiles a un pH elevado no serán recuperados adecuadamente (70-120%), según lo estipulado por el documento SANTE (European Commission. Directorate General for Health and Food Safety, 2017).
El utilizar el método QuEChERS AOAC 2007.01 (Lehotay, et al., 2005) permitió ampliar el número de analitos determinados en él, ya que la presencia del buffer acético-acetato formado in situ en el solvente de extracción hace que aquellos analitos que no se recuperaban en el método original sí puedan ser analizados adecuadamente con este agregado.
En la etapa de clean-up de tipo dispersivo se pueden emplear diferentes sorbentes en función de las características de la matriz en estudio. Se emplea PSA (amina primaria/secundaria) para la eliminación de ácidos orgánicos y pigmentos polares y RP-C18 para la eliminación de la mayor parte de los lípidos y esteroles presentes en las matrices con alto contenido graso.
Materiales y métodos
Reactivos y materiales
Acetonitrilo (MeCN) y Acetato de etilo (AcOEt) calidad HPLC fueron comprados a Pharmco (Brookfield, CT, USA). Sulfato de magnesio anhidro (MgSO4), Acetato de sodio anhidro (NaAc), Amina primaria y secundaria (PSA) y RP-C18 fueron obtenidos de Scharlab S.L (Barcelona, España).
Los estándares de pesticidas de alta pureza fueron obtenidos de Dr. Ehrenstorfer (Augsburg, Alemania) y guardados a -20 °C. Las soluciones madre individuales de cada pesticida (2000 mg L-1) fueron preparadas en AcOEt y guardadas en viales de vidrio color ámbar con tapa rosca y contratapa de teflón y en ausencia de luz a -20 °C. Los mix de soluciones stock utilizados para las calibraciones y las fortificaciones fueron preparados a partir de las soluciones stock individuales mediante diluciones adecuadas.
Instrumentos
Los residuos de pesticidas se analizaron usando cromatografía de gases acoplada a espectrometría de masas (GC-QqQ-MS). El equipo empleado fue un GC 2010 Ultra acoplado a un espectrómetro de masas triple cuadrupolo TQ8040 (Shimadzu).
El volumen de inyección fue de 1 μL de cada muestra en modo splitless y se usó un autosampler AOC 20 i+s. Se empleó una columna capilar RXi-5MS Sil (5% difenil/ 95% dimetil polisiloxano, 30 m; 0,25 mm d.i; 0,25 µm film) de Restek (Bellefonte, PA, USA). La temperatura del inyector fue 280 °C, el gas portador empleado fue Helio de alta pureza a un flujo de 1 mL min-1. El programa de temperatura del horno se describe en la Tabla 1.
Tabla 1. Condiciones del programa de temperatura del horno del cromatógrafo de gases empleado en el estudio.

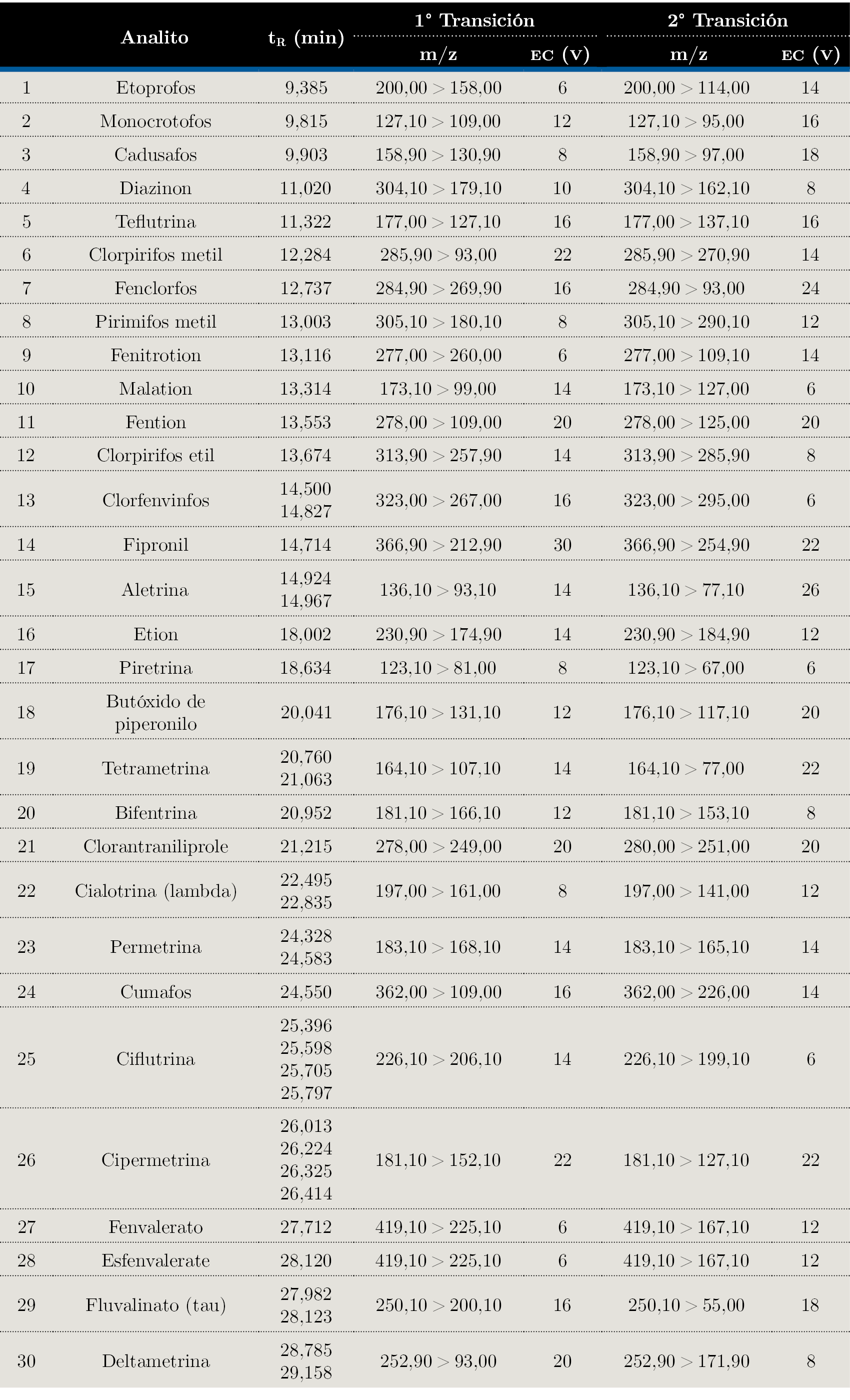
La temperatura de la interfase fue 300 °C y la temperatura de la fuente de ionización fue 230 °C. Se trabajó con un voltaje del detector de 1,25 kV y se empleó Argón (200 kPa) como gas de colisión. En todos los experimentos se operó en modo de Monitoreo de Reacciones Múltiples (MRM), ajustando las energías de colisión (EC) y empleando Smart Pesticide Database (SPDB) y MRM Optimization Tool (Restek, Bellefonte, PA, USA), como se describe en la Tabla 2.
Tabla 2. Optimización por GC-QqQ-MS de los 30 analitos evaluados en este estudio.
tR: tiempo de retención; EC: Energía de colisión expresada en V.
Selección de los analitos de interés y validación de la metodología
La selección de los analitos del estudio se hizo en base a un análisis de mercado y a la lista de importación de fitosanitarios del MGAP 2012-2017 (Ministerio de Ganadería Agricultura y Pesca, s.d.) para uso en ganadería. Se seleccionaron un total de 30 activos entre piretroides (Py) y organofosforados (OP).
Muestras
Las muestras de leche empleadas para el desarrollo y validación del método fueron obtenidas de un tambo del departamento de Artigas y guardadas en freezer a -18 °C hasta su estudio. El tiempo máximo de conservación de la muestra en freezer previo a su análisis fue de 45 días.
Preparación de muestra
Extracción
Para la extracción se siguió el esquema de QuEChERS AOAC 2007.01 (AOAC International, 2016) para matrices con alto contenido en grasa (Lehotay, et al., 2005).
Se homogeniezó la muestra a temperatura ambiente (25 °C) y en shaker mecánico durante 10 minutos, se pesaron (10,00 ± 0,05) g de leche cruda en un tubo de polipropileno de 50 mL. Se agregaron 10 mL de MeCN (1% HAc) y se agitó manualmente durante 1 minuto.
Se agregaron 4 g de MgSO4 y 1 g de NaAc y se agitó vigorosamente durante 1 minuto. Se centrifugó durante 5 minutos a 1400 RCF.
Clean-up dispersivo (d-SPE)
Se tomó una alícuota de 4 mL del sobrenadante y se colocó en un tubo de polipropileno de 15 mL con 200 mg PSA, 200 mg RP-C18 y 600 mg MgSO4.
Se llevaron los tubos a un vortex y se agitó durante 30 segundos a 1000 RCF. Posteriormente se centrifugaron los tubos durante 5 minutos a 1400 RCF. Se tomaron 2 mL del sobrenadante purificado, se colocaron en tubos de vidrio y se evaporó el solvente empleando corriente de N2 en un Turbovap® con un baño de agua a 45 °C.
Se retomó el residuo en 1,00 mL de AcOEt calidad HPLC, se filtró el extracto empleando filtros de jeringa de PTFE de 0,45 µm y se transfirió a un vial color ámbar de autosampler para su inyección en GC-QqQ-MS.
Validación de la metodología
Se validó la metodología de acuerdo con los lineamientos del documento SANTE/11831/2017 de la Unión Europea (European Commission. Directorate General for Health and Food Safety, 2017), en el cual se establece una serie de procedimientos analíticos y su validación para el análisis de residuos de pesticidas en alimentos y piensos. Se describen abajo las cifras de mérito evaluadas.
Veracidad
Se evaluó la veracidad del método a través de los porcentajes de recuperación para cada analito a cinco concentraciones: 5; 10; 20; 50 y 100 µg kg-1 (n=5 para todos los casos). Para realizar las recuperaciones se empleó una muestra blanco a temperatura ambiente y se adicionaron los volúmenes apropiados de las soluciones stock de los pesticidas seleccionados para lograr las concentraciones de trabajo. Las fortificaciones se agitaron empleando Vortex durante 1 minuto a 1000 RCF y se dejaron reposar durante 15 minutos antes de comenzar los análisis.
Precisión
La precisión se evaluó a través del grado de dispersión de las recuperaciones a cada concentración de trabajo. Se evaluó a través de la repetitividad (dispersión de los resultados de un batch de trabajo en un período corto de tiempo) y de la precisión intermedia (dispersión de los resultados de un batch de trabajo realizado en diferentes días por tres diferentes analistas).
Linealidad
La linealidad se evaluó en solvente y en matriz (matrix matched calibration). Se analizaron coeficientes de determinación (R2) y “back-calculated concentration” (BCC); además se realizó una inspección visual para cada caso.
Efecto matriz
El efecto matriz se evalúo como la relación entre la sensibilidad analítica de la curva preparada en matriz en relación a la sensibilidad de la curva de calibración preparada en solvente.
Se calculó el efecto matriz expresado en forma porcentual empleando la Ecuación 1.
$$EM\thinspace(\%) =\left[\left({pendiente\thinspace CCM\over pendiente\thinspace CCS}\right){-1}\right]\times100$$ (1)
CCM: curva de calibración en matriz; CCS: curva de calibración en solvente.
Resultados y discusión
Estudios preliminares
Al inicio de este trabajo se plantearon tres alternativas para la determinación de ectoparasiticidas lipofílicos en leche cruda. El contenido de grasa en la matriz de trabajo dificulta la extracción de compuestos con Kow elevados, entre ellos los que pertenecen a las familias de los piretroides y los organofosforados.
Se planteó el esquema QuEChERS adaptado por Lehotay para matrices con elevado contenido en grasa (Lehotay, et al., 2005). Particularmente se realizó un desgrasado con hexano y luego un freeze-out de 12 horas. La propuesta del desgrasado con hexano se planteó como alternativa para poder remover el elevado contenido de grasa presente en la leche cruda, donde los ácidos grasos se particionan hacia la fracción hexánica permitiendo una mejor limpieza del extracto de MeCN. Se completó la reducción a un mínimo del contenido en lípidos en el extracto empleando la metodología denominada freeze out, que consiste en precipitar las grasas a bajas temperaturas. Se dejaron en este experimento los tubos en freezer a -18 °C durante 12 horas y se tomó de la fase orgánica para continuar con el esquema del clean-up dispersivo propuesto por Lehotay et al. (2005).
La comparación de las tres metodologías a través de la evaluación de los porcentajes de recuperación al nivel de 100 µg kg-1 arrojó resultados favorables hacia la primera opción, por lo que se eligió el método QuEChERS para continuar con la validación de la metodología.
Validación de la metodología
Se realizaron recuperaciones a cinco concentraciones diferentes, por quintuplicado (n=5) para todos los casos. Los valores de recuperaciones y las desviaciones estándares relativas expresadas en forma porcentual se muestran en la Tabla 3.
Tabla 3. Porcentajes de recuperación (Rec) a las cinco concentraciones de trabajo y sus desviaciones estándares relativas (RSD) asociadas.
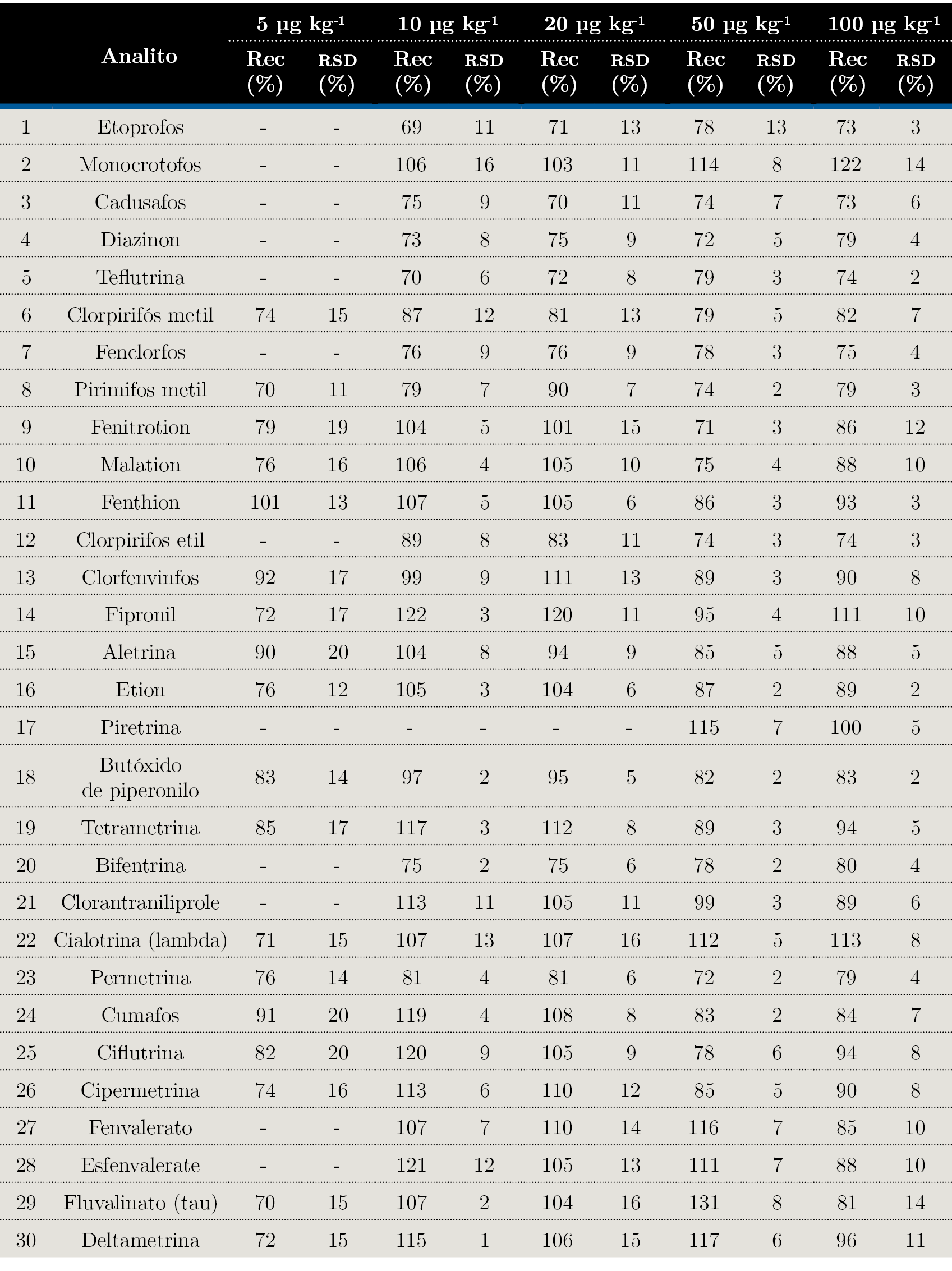
Se pudo comprobar que el método es veraz a todos los niveles de trabajo ya que los porcentajes de recuperación de los compuestos evaluados se encontraban entre (70-120) %. En casos como el del Etoprofos a 10 µg kg-1, el porcentaje es inferior al rango de aceptación (69%) pero con una desviación estándar relativa pequeña, por lo que el compuesto podría ser incluido en el método de análisis de rutina.
Al nivel más bajo, el 60% de los analitos se recupera con un porcentaje aceptable y cabe destacar que todos los analitos tienen al menos dos niveles de concentración en los que los criterios de veracidad y precisión se cumplen según los lineamientos de SANTE 2017 (European Commission. Directorate General for Health and Food Safety, 2017).
Respecto a la precisión, el grado de dispersión de las muestras expresado en la Tabla 3 como repetitividad fueron para todos los casos menores o iguales al 20%.
El ensayo de precisión intermedia se realizó con tres analistas evaluando todos los niveles de concentración trabajados. Los resultados obtenidos expresados como RSD (%) no superaron el 20%.
Para los estudios de linealidad se construyeron curvas de calibración con seis concentraciones en cada caso, tanto en solvente como en matriz.
Pese a que la guía SANTE (European Commission. Directorate General for Health and Food Safety, 2017) no exige inspección visual y evaluación de los coeficientes de determinación, se realizaron las curvas para cada analito de la respuesta analítica en función de la concentración (expresada en µg de pesticidas por kilogramo de leche). La Figura 1 muestra un gráfico de linealidad para Cipermetrina y su cromatograma.
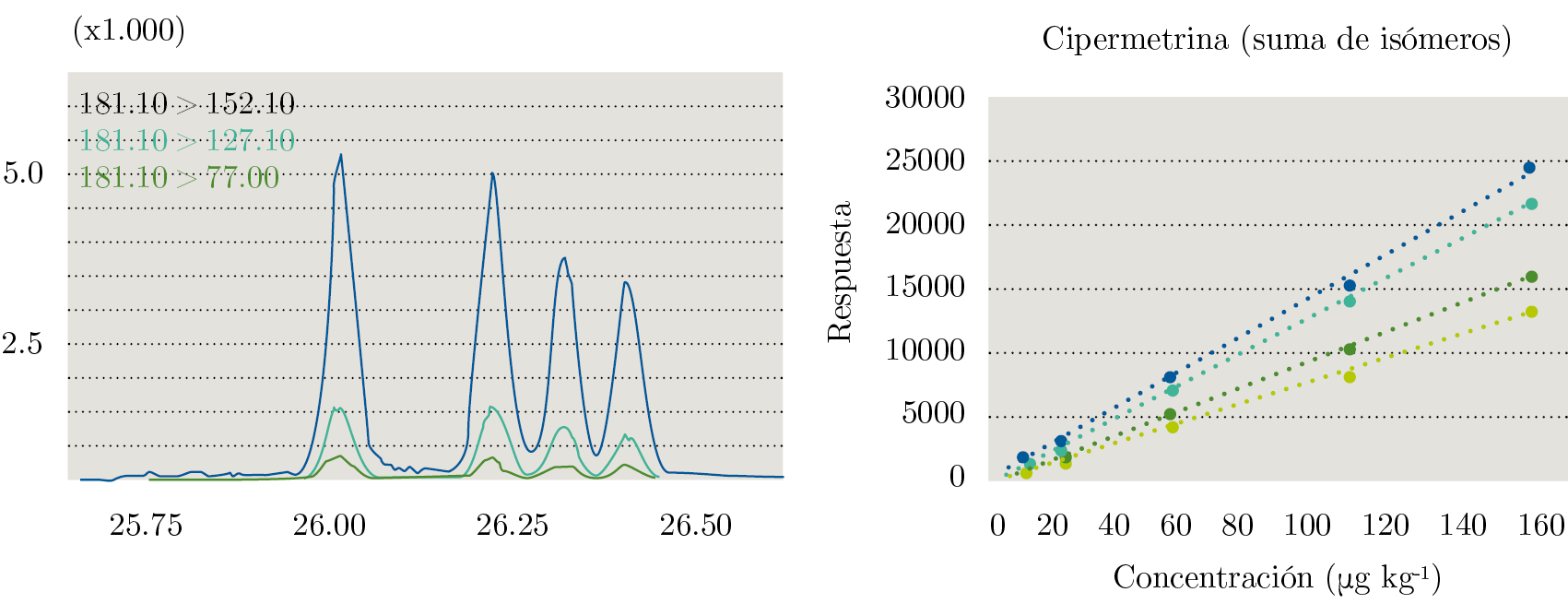
Figura 1. A la izquierda, cromatograma de Cipermetrina. En el eje de las abscisas se grafica el tiempo (en minutos) y en el eje de las ordenadas, la respuesta analítica. A la derecha, curva de calibración en matriz (matrix matched calibration) para sus cuatro isómeros por separado. Para todos los isómeros se grafica concentración del pesticida en función de la respuesta analítica.
Para todos los casos se observa comportamiento lineal con R2 superiores a 0,99. Se realizaron los cálculos de “Back Calculated Concentration” (BCC) como se muestra en la Ecuación 2.
$$BBC\thinspace(\%) ={(Cmedia-Creal)\over Creal}\times 100$$ (2)
Para todos los casos, tanto en solvente como en matriz, los porcentajes de BCC fueron ± 20% cumpliendo con los requisitos establecidos por SANTE (European Commission. Directorate General for Health and Food Safety, 2017).
De esta manera, el rango lineal para todos los analitos fue de (2,5-150) µg kg-1.
Empleando la información obtenida del estudio de la linealidad (pendientes de las curvas de calibración), se utilizó la Ecuación 1 y se evaluó el efecto matriz en forma porcentual.
Todos los compuestos presentan un efecto de aumento de la señal en matriz con respecto a la calibración en solvente, excepto clorantraniliprole, que presenta supresión de señal.
De los 30 compuestos evaluados, 21 presentan un efecto matriz bajo, es decir, el valor numérico obtenido de la Ecuación 1 (en módulo) se encuentra entre (0-20) %. Ocho compuestos presentan un efecto matriz moderado (20-50) % y solamente un compuesto presenta un efecto matriz elevado (>50 %). Esos resultados se pueden observar en la Tabla 4.
Tabla 4. Datos obtenidos para el estudio de linealidad y efecto matriz.
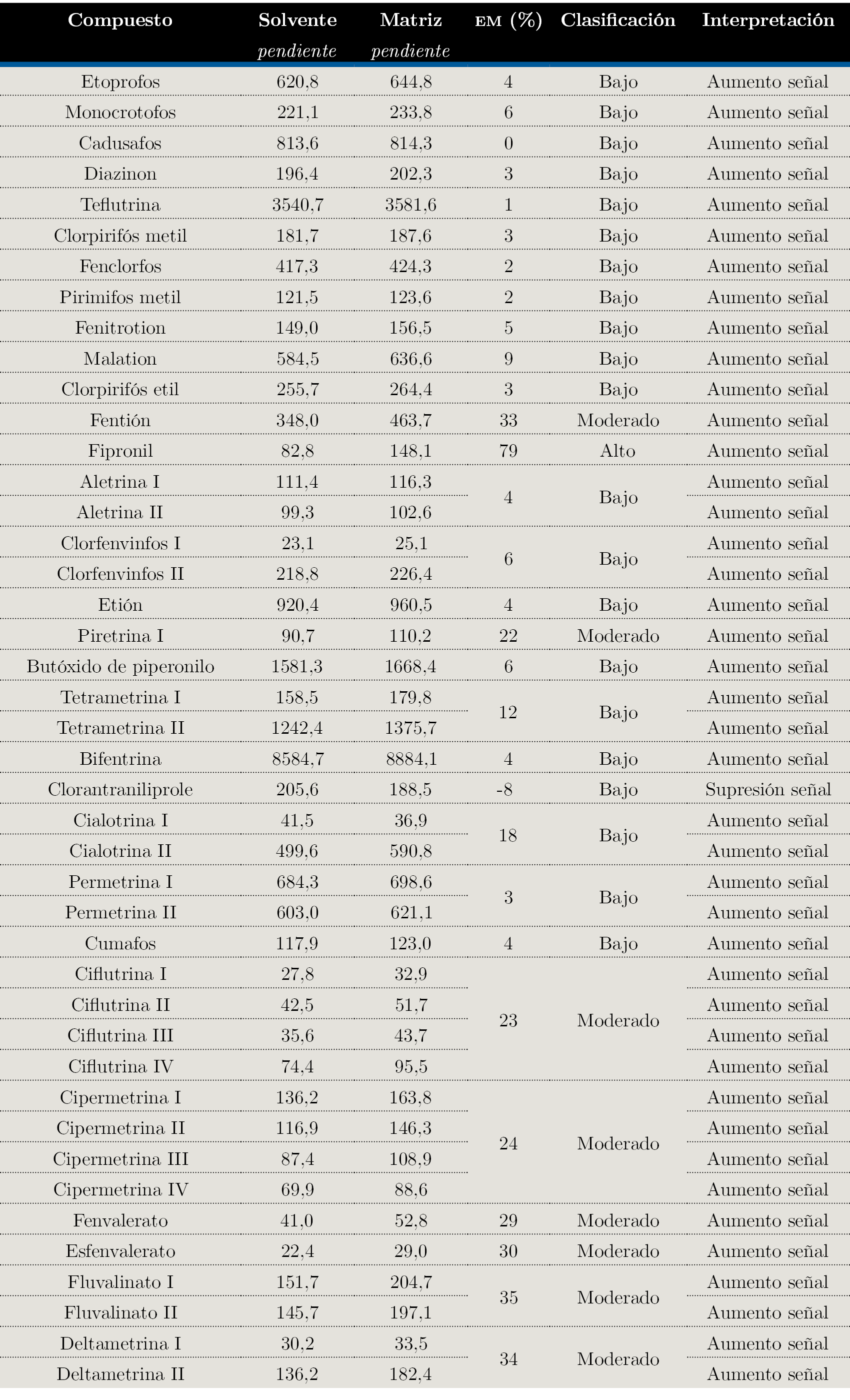
Pend: pendiente de la curva de calibración; EM: Efecto matriz calculado empleando la Ecuación 1 y expresado en forma porcentual.
Se puede observar en la Figura 2 la comparación de los gráficos de las curvas de calibración en solvente y matriz para Fipronil. Fipronil fue el único compuesto que presenta un EM positivo pronunciado (+79 %).
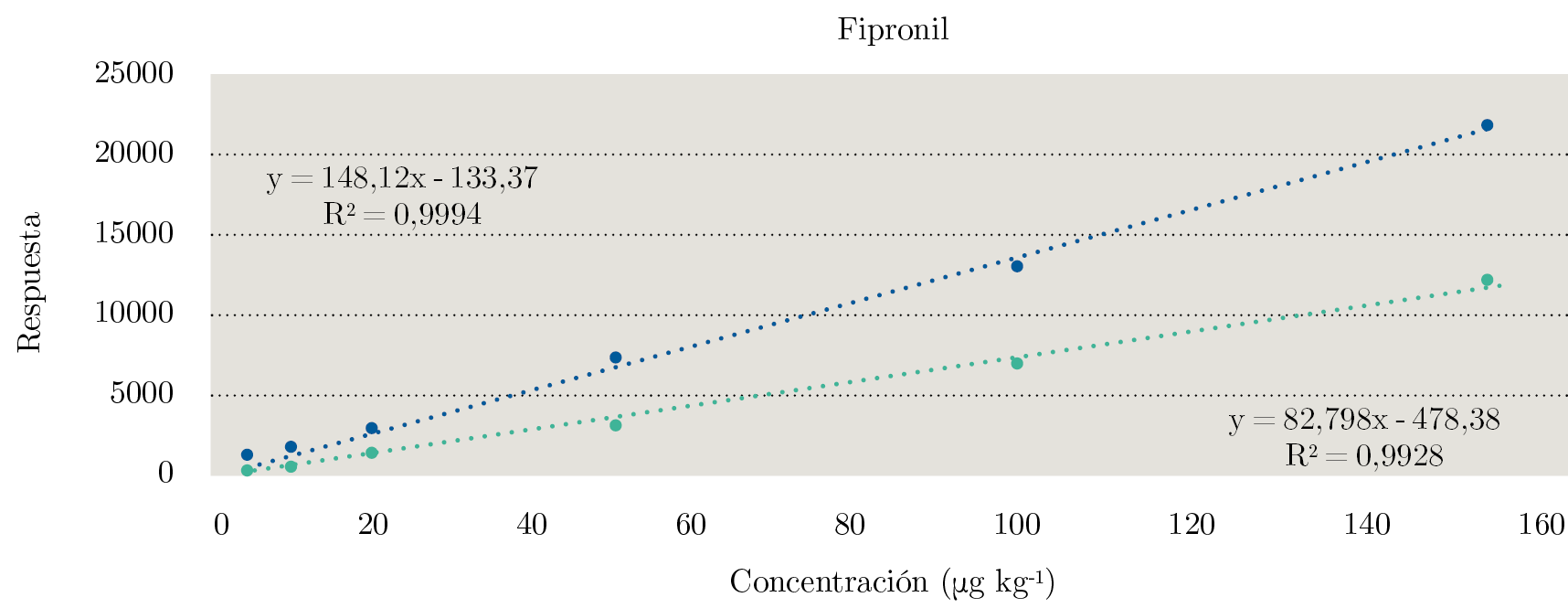
Figura 2. Comparación de las curvas de calibración realizadas para Fipronil. En verde, curva de calibración en solvente, y en azul, curva de calibración en matriz.
Respecto a los límites de cuantificación, se tomó como criterio establecerlo como el nivel más bajo de concentración al que se cumplen criterios de veracidad y precisión (recuperaciones entre 70-120% con RSD’s < 20%). Como se puede observar en la Figura 3, el límite para la mayoría de los compuestos es 5 µg kg-1. Solamente piretrina presenta un LOQ de 50 µg kg-1.
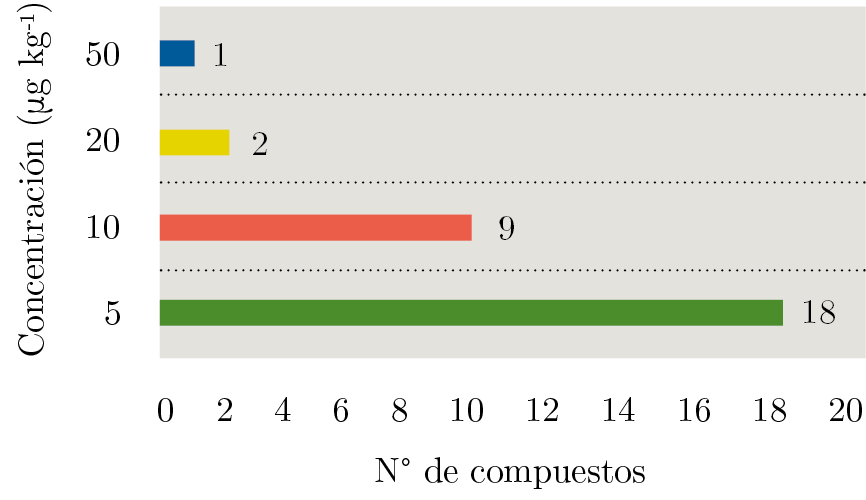
Figura 3. Límites de cuantificación (LOQ) expresados en µg kg-1 para los diferentes compuestos en estudio; 18 compuestos presentaron un LOQ de 5 µg kg-1 (barra verde), nueve compuestos presentaron un LOQ de 10 µg kg-1 (barra naranja), dos compuestos un LOQ de 20 µg kg-1 y solamente un compuesto un LOQ a 50 µg kg-1.
Conclusiones
Se ajustó y validó una metodología multirresiduo para el análisis de ectoparasiticidas lipofílicos (piretroides y organofosforados) en leche cruda empleando un método sencillo, rápido y que demostró a través de los datos de la validación ser adecuado para su propósito.
La metodología validada es una herramienta útil para determinar curvas de decaimiento de los compuestos aplicados de manera de asegurar a la leche su calidad e inocuidad alimentaria.
Este método se emplea rutinariamente en el Laboratorio de Análisis de Residuos de Pesticidas (GACT) en Facultad de Química (UdelaR) para la determinación de ectoparasiticidas en leche vacuna.
Reconocimientos
Los autores agradecen a la Red Analítica de Laboratorios de América Latina y el Caribe (RALACA-IAEA) por proveer los estándares para el estudio, a Dexin (Grupo Químico SRL) y SHIMADZU Brasil (GC-Team: R. Kitamura y F. Róveri) por brindar el soporte de software e instrumental del equipo empleado.